
INTRODUCTION
The hypotonia involves a reduction in muscle tone, which hinders passive mobilization and can affect muscle strength [4]. Symptoms can range from impaired consciousness to feeding problems, seizures, and abnormal postures [3]. Identifying the underlying cause can be complicated and costly, especially since many cases do not have a clear cause, resulting in a medical challenge [4].
Genetic disorders are responsible for 60% of hypotonia cases, while about 80% of congenital hypotonias are of central origin, with hypoxic-ischemic encephalopathy being the most common cause, followed by other conditions including genetic syndromes [3] such as that associated with variants in the PLA2G6 gene. Variants of the PLA2G6 gene are linked to 3 serious neurological diseases. One of them is Infantile Neuroaxonal Dystrophy (INAD), a severe progressive neurodegenerative disease [5] that usually manifests before the age of 3 years [6,7]. They are also linked to Atypical Neuronal Dystrophy (ANAD) or NBIA2B that can appear from the age of 3 to late adolescence, presenting symptoms such as ataxia, rigidity and mental deterioration, among others and variants associated with dystonia-parkinsonism of onset in adulthood (between 20 and 40 years of age), which is characterized by symptoms such as cognitive impairment and parkinsonian manifestations [7].
In 1952, Seitelberger reported for the first time that Infantile Neuroaxonal Dystrophy (INAD), a disease with an autosomal recessive inheritance mechanism, manifested with psychomotor regression, neurological regression and hypotonia that progressed to spastic tetraparesis and vision impairment. Symptoms begin between 6 months and 3 years, and usually result in death before age 10 due to complications such as aspiration pneumonia. Currently, there is no curative treatment for INAD, only supportive therapies are available to relieve symptoms and prevent complications [8]. The prevalence of this entity is unknown, classifying it as an ultra-rare neurodegenerative disorder [9]. In the context of these diseases, reverse phenotyping becomes important to establish genotype/endotype/phenotype correlation. This has the benefit of broadening the understanding of genetic diseases, confirming new disease-genotype relationships, and analyzing rare phenotypes in more detail [10].
Next, we describe a clinical case of an older infant with progressive neuromuscular manifestations with no known related family history, non-consanguineous parents in which a specific diagnosis was reached through the use of genomic technique, and a genotype/phenotype correlation could be established, with a medical condition of very low population prevalence, in order to establish directed and individualized medical actions, preventive, anticipatory measures, and create awareness among health professionals about the positive impact of specific diagnosis, comprehensive management, approaching precision, prediction, personalized, preventive, participatory medicine in order to be carried out at the population level.
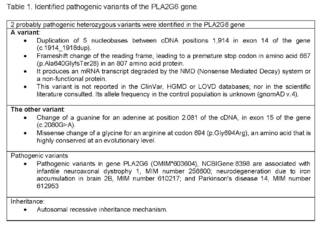
A specific genomic study was requested by NGS (Next Generation Sequencing) exome sequencing and CNV (Copy Number Variants) analysis for genes associated with congenital hypotonia (1,621 genes). The study identified two probably pathogenic heterozygous variants in the PLA2G6 gene.
CLINICAL CASE
The 2-year-old patient, the product of first-time parents in the third decade of life, not consanguineous, with full-term birth and without complications. No prenatal ultrasound alterations, no exposure to toxins or teratogens. Anthropometric measurements appropriate for gestational age. No family history of genetic diseases or congenital malformations. It presents with severe global neurodevelopmental delay and regression of milestones acquired at the age of one year and four months.
From birth he presents generalized hypotonia, left horizontal nystagmus and strabismus. Phenotype related to prominent forehead, epicanthal folds, retrognathia, right ear over the implantation limit, spaced and irregular teeth, axial hypotonia, does not achieve cephalic support, does not crawl, does not bidepose or walk, although it smiles and looks for the sound source, does not convulse, and feeds by mouth.
Patient with a syndromatic diagnosis of hypotonia of central axial origin with metabolic studies in order to rule out inborn error of metabolism -mitochondriopathy such as venous gases, ammonium, lactic acid, lactate/pyruvate, carnitine profile, acyl-carnitines, organic acids, fatty acids, creatine-phosphokinase, ketones, glycemia, amino acid measurement, transaminases, urinalysis and normal anion-gap; brain magnetic resonance imaging that reports cerebellar atrophy.
For this reason, given the complexity of the clinical manifestations, high risk of de novo genetic disease vs.autosomal recessive inheritance, a genomic study directed exome sequencing by NGS (New Generation Sequencing) + CNV (Copy Number Variants) was requested for genes related to congenital hypotonia (1,621 genes), which identified 2 heterozygous variants probably pathogenic in the PLA2G6 gene (Table 1).
DISCUSSION
The PLA2G6 gene is identified as the only gene committed so far to the generation of INAD, it is located on chromosome 22 in 22q13.1. It was first described as an INAD-related gene in 2006. PLA2G6 encodes group VI calcium-independent phospholipase A2, an enzyme that plays an important role in cell membrane homeostasis.In variants that alter the PLA2G6 gene, a failed repair of oxidative damage to phospholipid membranes occurs, leading to changes in membrane permeability [8,11]. According to the artificial intelligence genomic platform Mastermind (https://mastermind.genomenon.com/) it is confirmed that phospholipase A2 catalyzes the release of fatty acids from phospholipids and may play a role in the release of arachidonic acid, in the remodeling of phospholipids, the synthesis of leukotrienes and prostaglandins, fas-mediated apoptosis and transmembrane ion flow in glucose-stimulated B cells.
The PLA2G6 protein is also associated with the maintenance of mitochondrial integrity and function, and has been implicated in the regulation of autophagy, a cellular degradation pathway important for cell survival and homeostasis. Several transcription variants encoding multiple isoforms have been described, but to date only the full nature of three of them has been determined.When using this Mastermind platform, PVS1 (ClinGen haploinsufficiency score 30; DECIPHER haploinsufficiency score (pHaplo score) 0.29; ClinVar (#P/LP LOF variants) / (all P/LP variants) for this gene 27.3%; ClinVar (# of P/LP LOF + CNV) / (# all P/LP) 31.6%; LOEUF from gnomAD6 0.86.- PP2 (ClinVar (P/LP missense)/(all P/LP) for this gene 57.80%.
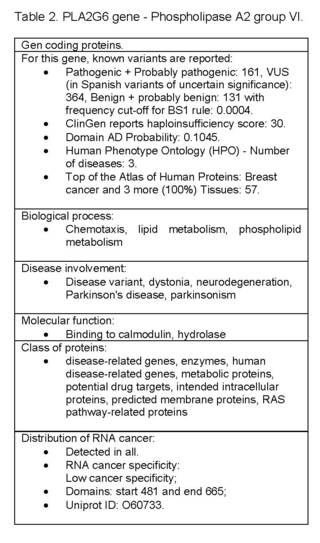
PLA2G6 - phospholipase A2 group VI, protein-coding gene, known variants are reported: Pathogenic + Probably pathogenic: 161, VUS (in Spanish variants of uncertain significance): 364, Benign + probably benign: 131 with frequency cut-off for BS1 rule: 0.0004 (Figure 1).ClinGen reports haploinsufficiency score: 30.Domain AD Probability: 0.1045.Human Phenotype Ontology - HPO (Human Phenotype Ontology) - Number of diseases: 3.Top of the Atlas of Human Proteins: Breast cancer and 3 more (100%) Tissues: 57.
Biological Process: Chemotaxis, Lipid Metabolism, Phospholipid Metabolism; Disease involvement: disease variant, dystonia, neurodegeneration, Parkinson's disease, parkinsonism; Evidence of Hpa Evidence at the protein level; Molecular function: binding to calmodulin, hydrolase; Protein class: disease-related genes, enzymes, human disease-related genes, metabolic proteins, potential drug targets, intended intracellular proteins, intended membrane proteins, RAS pathway-related proteins; Distribution of RNA cancer: detected in all; RNA cancer specificity Low cancer specificity; Domains: start 481 and end 665; Uniprot ID: O60733 (Table 2).
In the case presented, 2 heterozygous variants of clinical significance were identified, probably pathogenic in the PLA2G6 gene, identified by genomic study: Exome sequencing NGS + CNV technology in genes associated with congenital hypotonia.
Pathogenic variants in gene PLA2G6 (OMIM*603604), NCBIGene: 8,398 are associated with Childhood Neuroaxonal Dystrophy 1, MIM number 256600; Neurodegeneration with iron accumulation in the brain 2B, MIM number 610217; and Parkinson's Disease 14, MIM number 612953; all with an autosomal recessive inheritance mechanism.
INAD manifests between 6 months and 3 years of age with rapid motor and cognitive deterioration or neurological regression [12,13]. Early truncal hypotonia progresses to tetraparesis, usually spastic, with total loss of voluntary muscle control; visual symptoms, such as strabismus, pendulum nystagmus, uncoordinated eye movements, optic atrophy, and vision loss.Some cases present with seizures [13,14].
The course of the disease ends in mortality in the first decade. Most of them have cerebellar atrophy appreciable on magnetic resonance imaging with T2 hyperintensity, being the earliest feature of the disease, they may also present elevated cerebral iron or optic atrophy, show spheroid bodies in the central nervous system and pathological inflammation of the axons [8,12]. Cerebral iron accumulation, with hyperintensity of the globus pallidus isolated in T2, is a late neuroradiological marker that occurs in 50% of cases, however, it is very specific [12]. The diagnosis of INAD should be suspected in individuals with the clinical features and MRI findings mentioned at a relatively young age, but eventually the diagnostic confirmation is given by genetic testing [14], mainly confirmation of PLA2G6 status, along with associated clinical phenotypes [15].
In a case series of 28 patients with INAD in 2020 (16 patients from Riyadh, Saudi Arabia, 8 patients from North and South America, and 4 patients from Europe found that speech impairment and loss of gross motor landmarks were the most common signs of the disease, nystagmus occurred in 60.7%, seizures in 42.9%, gastrointestinal disease in 42.9%, skeletal deformities in 35.7%, and strabismus in 28.6% [9].In 2021, in Iran, a case series was carried out with 3 pediatric patients with INAD, these showed hypotonia and psychomotor regression before the age of three, followed by spastic characteristics. One developed cone dystrophy and another seizures and abnormal EEG patterns, uncommon findings in INAD [6]. In another study published in 2023 that included 18 cases of INAD, gross motor regression was found to be the most frequent initial symptom and cerebellar atrophy was the most prevalent brain imaging finding, observed in more than 50% of individuals [16]. In the case described, the manifestations begin before 3 years of age, associated with severe neurodevelopmental delay with progressive neurological regression, axial hypotonia, visual alterations such as nystagmus and strabismus, these being early characteristics of the disease; and in the images of the MRI with cerebellar atrophy.The diagnosis was confirmed by current genomic techniques (NGS and CNV) with detection of genes related to congenital hypotonia and finding of a compound heterozygous variant in the PLA2G6 gene, associated with childhood neuroaxonal dystrophy (INAD).
In the Orphanet database, it is referenced that the prevalence of INAD is unknown, with a description of more than 150 cases, most of which are classic INAD.In the Human Gene Mutation Database (HGMD), more than 200 variants have been recorded in PLA2G6.A total of 218 variants have been reported in PLA2G6 and more than 130 variants that cause INAD have been described [8]. Innovative therapies such as enzyme replacement and gene therapy for INAD are in early stages of development, which means that available treatments remain palliative [15] and supportive, encompassing spasticity management, seizures, and enteral tube feeding or gastrostomy, to prevent bronchoaspiration.Iron chelation therapy is not recommended.The progression is very rapid, severe spasticity, progressive cognitive impairment and visual impairment can lead to a vegetative state [14].
The parents of someone affected can be carriers (obligate heterozygotes) of a pathogenic PLA2G6 variant without presenting symptoms. Each sibling of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of not being affected or a carrier.In these situations, genetic counseling is crucial to help families make informed medical and personal decisions [17].
A search for this variant was carried out in artificial intelligence assistants.According to GenAI's first genetic variant assistant, VarChat (https://varchat.engenome.com/) which for the genomic variant p.Gly694Arg reports that this variant is not described in the literature and represents a missense mutation in the PLA2G6 gene, where glycine at the amino acid 694 position is substituted for arginine.This change of a single amino acid is caused by a point mutation at the nucleotide level, which alters the codon from one that codes for glycine (GGG/GGA/GGT/GGC) to one that codes for arginine (AGA/AGG/CGT/CGC/CGA/CGG). Substituting a small, nonpolar glycine residue for a larger, positively charged arginine residue can have significant implications for protein structure and function, potentially leading to altered enzyme activity, protein stability, or interactions with other molecules.
Dysregulation or mutations in PLA2G6 have been associated with a variety of neurodegenerative disorders, indicating their significant role in neural health and function. Therefore, the p.Gly694Arg variant could have implications for the normal enzymatic activity of PLA2G6 and could contribute to the pathogenesis of associated diseases. For the p.Ala640GlyfsTer28 variant in the PLA2G6 gene, it is reported that it is not described in the literature.The reported variant is a frameshift mutation that results from an insertion or deletion that alters the gene frame.
The designation 'p.Ala640GlyfsTer28' indicates that the alteration occurs at the amino acid 640 position, where an alanine (Ala) is expected to be replaced by a glycine (Gly), and this change leads to a reading frame change. The 'fs' denotes that the frameshift mutation will result in a series of 28 aberrant amino acids before encountering a premature termination codon (Ter). This premature stop codon is predicted to result in a truncated protein product or could potentially trigger missense mRNA-mediated degradation, leading to reduced levels of the protein PLA2G6. The p.Ala640GlyfsTer28 variant in the PLA2G6 gene likely has a detrimental effect on protein function, contributing to the pathogenesis of these aforementioned neurodegenerative diseases.
These variants are not reported in the databases described, nor in the literature, with allelic frequency in unknown control populations, together with knowledge about functionality, biological bases, haploinsufficiency, molecular factors, protein structure and function, functional studies, use of artificial intelligence tools and according to Richards, et al.Standards and guidelines for the interpretation of sequence variants.2015 [18], American College of Medical Genetics and Genomics, Association for Molecular Pathology, ClinGen, these variants are classified as probably pathogenic, which is why genotype/endotype/phenotype correlation can be established; associated with the importance of understanding the phenotypic-genotypic heterogeneity and possible probabilities of expression of other medical conditions related to deleterious variants in this gene.
CONCLUSIONS
The neuromuscular diseases are considered a medical challenge and an important cause of pediatric morbidity and mortality, they are related to progressive disability, a significant impact on the quality of life of those who suffer from them and their families, so reaching a specific diagnosis is essential.Congenital hypotonia is linked to a number of disorders such as INAD, which makes it difficult to make a specific diagnosis, hence the importance of the implementation of current genomic diagnostic tests, which confirm the condition, in order to establish a defined therapeutic plan, take preventive measures, avoid complications and improve the well-being of both the patient and their family.
Reverse phenotyping is important in these diseases, improving genetic understanding, validating new genotype-endotype-phenotype associations, and exploring uncommon phenotypes.In addition to its usefulness in anticipatory preventive medicine, especially in cases with phenotypic heterogeneity such as this gene, in which continuous monitoring is important.This will allow us to determine if the gene manifests differently at different stages of the patient's life. The contributions of genomics are significant in addition to helping to understand complex diseases and establish their diagnosis, reducing the number of unnecessary procedures and treatments, alleviating the emotional burden of confirming a diagnosis, guiding the performance of family genetic testing, facilitating genetic counseling and family planning, providing insight into the severity of the prognosis, and helping to determine the most effective treatments for the sake of a healthy and healthy approach, hyperpersonalized medicine, especially in these medical conditions with a very low population prevalence.
ETHICAL RESPONSIBILITIES
Protection of people and animals: The authors declare that, for the elaboration of this project, no experiments were carried out with humans or animals.
Data confidentiality: The authors declare that they have followed the protocols of the patient's home institution on the publication of data; the document does not contain data that would allow the identification of the patient.
Right to privacy and informed consent: The authors declare that they have obtained the informed consent signed by the patient's guardian.
ABOUT THE AUTHORS
Jenny Adriana Morán Fernández
She is a Pediatrician at the Universidad Libre seccional Cali, Colombia, and part of the Pediatrics Research Group (GRINPED).
Lina Johanna Moreno Giraldo
She is a pediatric physician, with a master's degree and doctorate in Medical Genetics. Attached to Universidad Libre Seccional Cali, Colombia. She is part of the Pediatrics Research Group (GRINPED), and Leader of the Neurogenetics and Metabolic Diseases Research Line (NEUROMET).