
Chromoanagenesis and Erythroderma as diagnostic clues in a critically ill patient

 ,
, Authors
DOI:
https://doi.org/10.37980/im.journal.ggcl.20242414Keywords:
chromoanagenesis, cancer, oncogenetics, lymphoproliferative process, erythroderma, paraneoplasiaAbstract
We report the case of a patient with severe multisystemic failure in which massive chromosomal changes in mosaic state were detected, which are considered to be associated with the possible underlying pathology: a T-cell lymphoproliferative syndrome. The patient also presented a case of Erythroderma of unknown cause several years earlier, with a progressive decline in her physical condition. The spectrum of clinical manifestations is then presented as a nonspecific paraneoplastic cutaneous picture of several years of evolution and findings of chromoanagenesis in the last phase of a lymphoproliferative morbid process. The new techniques of genetic analysis make it possible to obtain data to clarify the diagnostic processes and to identify a serious disorganization of the genetic information considered to be responsible for neoplastic changes.
Case Report
This is a 69-year-old female patient, third child of non-consanguineous parents, and family history of a sister diagnosed with Systemic Lupus Erythematosus (SLE), who was in the Intensive Care Unit, presenting systemic disease and complications that required recurrent hospitalizations for three months, with progressive worsening. The patient presented evidence of hyperinflammation with increased Erythro-Sedimentation Rate (ESR) and C-reactive protein.
Chest CT revealed areas of pulmonary fibrosis and ground-glass images. She also presented bicytopenia with severe anemia and moderate thrombocytopenia, recurrent infections difficult to manage, anasarca, pleural effusion and progressive organ failure. The presence of multiple adenopathies led to suspect a lymphoproliferative syndrome, but the lymph node and bone marrow biopsies performed up to that moment had been negative for lymphoma, and only presented with a finding of moderate dysplasia of the white series, without other definitive conclusions.
In this critical state, and without an etiological diagnosis, an evaluation by Genetics was requested, who recommended performing a whole genome test looking for data related to oncogenetic alteration in the germ line, innate immunity or other predisposing genetic condition, since there were no specimens available for another type of molecular study. The patient died a few days later, with genetic test results pending.
Prior to the current clinical situation, the patient had a history of a condition that began approximately seven years ago, being evaluated by Hematology for the presence of a non-painful right retroauricular node, without B symptoms (fever, sweating, weight loss), nor other findings in laboratory and imaging tests, nor evidence of visceromegaly or other lymphadenopathies. In addition, the patient presented at that time with erythema on extensive surfaces of the body, including face, trunk and extremities (Figure 1), associated with generalized pruritus accompanied by burning sensation and allodynia, areas of desquamation on the feet and other parts of the body, and edema of the lower extremities. A diagnosis of erythrodermaemia was made, and although the most frequent causes were ruled out, the patient left the dermatology evaluation without a conclusion and followed alternative medicine treatments with partial improvement of the skin condition.
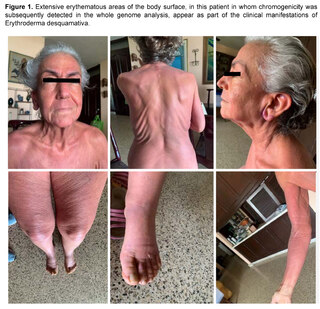
Six years later, the patient returned to the Hematology Department with lymphadenopathy in the neck, axilla, hepatosplenomegaly, history of recurrent candidiasis and persistent erythroderma, with the generalized edema, pleural effusion and progressive organ failure described above, with a prolonged period of hospitalization and the outcome described above.
Molecular Testing
From dried peripheral blood sample on filter paper, CentoGenome® MOx 1.0 Solo was performed: genomic DNA was enzymatically fragmented and labeled with Illumina-compatible adapter sequences. Libraries were sequenced from both ends (paired-end) on an Illumina platform to generate an average coverage depth of ~30x. A bioinformatics process based on Illumina's DRAGEN process as well as CENTOGENE's in-house bioinformatics process was applied. Reads were aligned to the Genome Reference Consortium Human Build 37 (GRCh37/hg19) genome assembly, as well as to the revised Cambridge Reference Sequence (rCRS) of human mitochondrial DNA (NC_012920). Sequence variants (SNVs/indels) and copy number variants (CNVs) were called using the DRAGEN algorithm, Manta and proper. All variants with a minority allele frequency (MAF) of less than 1% in the gnomAD database, and disease-causing variants reported in HGMD®, in ClinVar or in the CENTOGENE Biodatabank were evaluated..
Although the evaluation focused on coding exons and flanking intronic regions, the entire gene was interrogated for candidate variants with a plausible association with phenotype. All potential patterns of mode of inheritance were considered. In addition, family history and clinical information provided were used to evaluate the identified variants with respect to their pathogenicity and disease causation. Variants are classified into five classes (pathogenic, probably pathogenic, VUS, probably benign, and benign) following ACMG guidelines for variant classification in addition to ClinGen recommendations. All relevant variants related to the patient's phenotype were reported. Benign and probably benign variants are not reported.
For CentoGenome® MOx, if biochemical analysis is applicable, it is performed after detection of relevant variants by sequencing in specific genes. This improves the diagnosis of metabolic disorders, optimizes variant classification and helps to determine the final contribution to the phenotype; the list of enzyme activity assays and biomarkers can be obtained at www.centogene.com/mox. CNVs of uncertain significance with no apparent relationship to patient phenotype were not reported. Mitochondrial variants with heteroplasmy levels of 15% or greater are reported. For the detection of SNVs and indels in the regions selected for downstream analysis, a sensitivity of 99.9%, a specificity of 99.9%, and an accuracy of 99.9% are achieved. The CNV detection software has a sensitivity of more than 95%. CENTOGENE has established strict quality criteria and validation processes for variants detected by NGS.
Variants with poor sequencing quality and/or unclear zygosity were confirmed by orthogonal methods. Consequently, a specificity > 99.9% was guaranteed for all reported variants. Screening for repeat expansions was performed with the ExpansionHunter algorithm for the following genes: AR, ATN1, ATXN1, ATXN2, ATXN3, ATXN7, ATXN8OS, ATXN10, CACNA1A, CNBP, CSTB, C9ORF72, DMPK, FMR1, FXN, HTT, JPH3, NOP56, PABPN1, PHOX2B, PPP2R2B, PRNP and TBP.
The technical results of the repeat expansion screening were correlated with the clinical information provided. Any repeat expansion called and considered relevant to the phenotype would be confirmed by an orthogonal method. GBA1 screening was performed using the Gauchian algorithm to detect recombination events affecting the region spanning exons 9-11 (NM_000157.3), a region that has the highest homology to GBAP1. Any recombination events detected would be reported only when considered relevant to phenotype.
Spinal muscular atrophy (SMA) screening was performed using the SMN Caller algorithm to detect SMN1 gene copy number. Any CNV detected would only be confirmed by an orthogonal method and reported when considered relevant to phenotype. Uniparental disomy (UPD) screening was performed using an in-house Mendelian inheritance error (MIE) algorithm to detect regions of homozygosity (ROH) for known clinically relevant chromosomal regions (6q24, 7, 11p15.5, 14q32, 15q11q13, 20q13 and 20).
CentoArray®: The Infinium™ Global Diversity Array with Cytogenetics kit (Illumina) was used for this analysis. This kit contains ~1.8 million SNP markers distributed genome-wide, in particular for more than 4,800 disease-associated genes with 99.9% exonic coverage, allowing the detection of copy number variations (CNVs). Genomic DNA was amplified, fragmented and hybridized on the array according to the manufacturer's instructions. Results were analyzed using NxClinical software (BioDiscovery).
CNVs exceeding a size of 50 kb for losses and 200 kb for gains were reported. Losses identified below the given thresholds were only reported if a clear phenotypic correlation with affected genes was observed. Results were interpreted using DGV, DECIPHER, ClinGen databases and additional available resources. CNVs are classified into five classes (pathogenic, probably pathogenic, VUS, probably benign and benign) according to ACMG recommendations for variant classification. Benign and probably benign CNVs were not reported.
CNVs were evaluated based on the patient's reason for consultation for this test and the clinical information provided. Full reporting of heterozygous CNVs in the context of recessive disease is beyond the scope of intended use for this test. Therefore, carrier status may not be disclosed. Any clinical concerns for recessive disorders should be reported to the reporting laboratory for appropriate consideration.
In accordance with regulatory requirements in prenatal testing, CENTOGENE only reports pathogenic and probably pathogenic CNVs of high penetrance. This array allows the analysis of regions with absence of heterozygosity (AOH). The presence of AOH on multiple chromosomes may be compatible with inheritance from a common ancestor.
Results
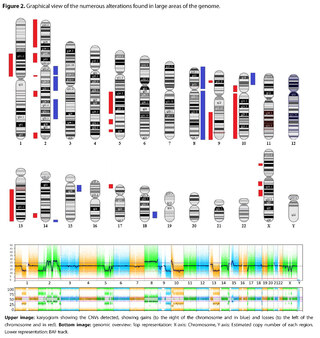
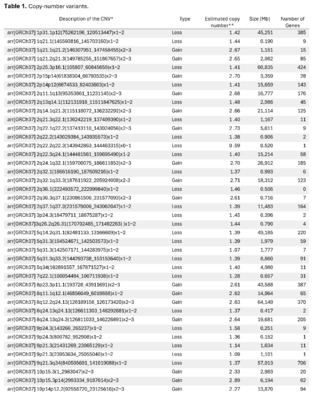
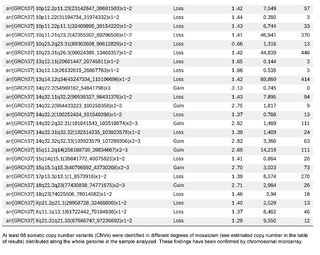
The presence of large copy number variants (CNVs) was identified, distributed throughout the genome in different degrees of mosaicism in the sample analyzed and, therefore, considered of somatic and non-germline origin (See Table 1) and confirmed by chromosomal microarray (See Figure 2), and which are compatible with a chromoanagenesis process, suggesting by the genetic laboratory to perform a hemato-oncologic evaluation, which in our context of clinical suspicion, would be a background lymphoproliferative disorder.
Discussion
The routine evaluation of the karyotype as a source of initial data to resolve clinical pictures, transmits us in the frequent image of chromosomes, the impression of stability of genetic information, in which alterations in number and structure are detected as sporadic findings. Only in bone marrow karyotypic evaluations is one more prepared to observe severe structural and numerical changes concurring in the same patient.
Although we know that this image of stability often does not correspond to the ultimate reality and that CGH (Comparative Genome Hybridization) techniques allow us to detect the presence of losses or gains of genetic material not detectable at the karyotype level and thus make the diagnosis of syndromes due to microdeletion or microduplication, the findings in the present case with the detection of a large-scale chromosomal debacle with numerous copy number variants (CNVs - Copy Number Variants), product of chromosomal rearrangement processes, is striking and unusual.
The term chromoanagenesis (chromosomal renaissance) was suggested by Holland and Cleveland in 2012 to describe a series of phenomena consisting of massive chromosomal reorganization processes, observed thanks to new technologies such as genomic sequencing and microarray used on this occasion [1]. Observed and described initially by Stephens et al [2], these real cataclysms in genetic information have been associated with neoplastic phenomena of different types and genetic-dysmorphic syndromes, although they have also been detected in the germ line of normal individuals, making them prone to a higher incidence of miscarriages and offspring with genetic syndromes [3].
Recall that genes control everything related to the design and functioning of the organism, including the control of cell growth, and that when these genes are altered they can lead to cancer. These genes participate through their products, which are proteins, in the inter- and intracellular communication system and that a quantitatively or qualitatively altered protein constitutes the initial mechanism for these disorders of cell growth control, since cancer is a genetic disease. Genetic damage is the so-called “necessary cause” and this damage can be produced by various mechanisms, called “sufficient causes” [4].
Apart from point mutations, chromosomal rearrangements can create fusion genes that produce “superproteins”: gain-of-function proteins that promote uncontrolled cell growth, or damage essential genes involved in DNA repair or tumor suppressor genes that monitor the occurrence of genetic damage and try to fix it, the latter two being loss-of-function events [5].
Chromoanagenesis, which includes three distinct mechanisms known as chromothripsis, chromoplexy and chromoanasynthesis, refers to processes of abrupt and cataclysmic remodeling of the genome that are triggered by different molecular circumstances, among which those related to cancer-related genes are involved. Chromotrypsis, defined by Pellestor as “a mutational event produced by multiple double-strand breaks recurrently in a single catastrophic event among a limited number of chromosomal segments and followed by reassembly of DNA fragments in a random order and orientation to form derivative chromosomes” [6], has been described as the subtype of massive chromosomal rearrangement present significantly in cases of cancer with chromoanagenesis, although, as we have mentioned, it can be found in other genetic disorders and even in the germline of healthy individuals, representing an increased risk of miscarriages or offspring with genetic disorders [7]. ”
On the other hand, chromoanasynthesis (chromosomal reconstitution) is another mechanism of chromoanagenesis whose effects are similar to those of chromothripsis, but which has been identified predominantly in syndromic genetic pictures with dysmorphias and intellectual disability [6,8,9], while chromoplexy (chromosomal rearrangement) could be the predominant mechanism in certain types of cancer in which chromoanagenesis is present, such as prostate cancer and others, and unlike chromothripsis, the rearrangements present few or no alterations in the number of copies [6].
All these phenomena seem to support the theory of cancer evolution based on one or few cataclysmic events or punctuated equilibrium theory [10], also hypothesized as one of the mechanisms of speciation in Nature, instead of the traditionally invoked model consisting of long-term cumulative or gradualist mutations, as Pellestor points out, although a mixed model has also been postulated in cancer cases [2,6].
Genetic information must be neither in excess nor in deficit, in order to guarantee the basic homeostasis of the cellular information system. The imbalance in a single gene is already capable of causing serious problems. In this case, the first of its kind reported in our country, the remarkable accumulation of complex chromosomal alterations is very striking, with 68 according to the report obtained. In order to realize the seriousness of the disorganization, it was evidenced that it involved 14 of the 23 chromosomes (61%). Of these changes, 66% were chromosomal losses and 34% gains. The most affected chromosomes were 2 (26%), 10 (13%) and 14 (10%). Gene losses ranged from 0 to 706 and gains from 0 to 387 (see Table 1).
Although we have no way of confirming this through additional studies, based on the description of the characteristics of the three mechanisms involved in chromoanagenesis and in which cases they occur, the changes presented in Table 1 would correspond to a chromothripsis phenomenon. In most cases, the severe genome disorganization that occurs in cases of chromoanagenesis, specifically of the chromothripsis type, would involve up to 2-4% of all cancers and have an ominous prognosis [6], which could be used in the future as a marker for the possible evolution of patients. However, there are no clear data on the level of tolerance of the system and on the resilience for recovery, and whether there is a threshold of degree of disorganization that, if exceeded, would mark a point of no return, even if the precise etiological treatment has been instituted.
Although there is no doubt that in the numerous cases of cancer reported with chromanoanagenesis, it is likely that there has been the concomitant presence of paraneoplastic symptoms, we have not found reports of patients in which Erythroderma is described as a process associated with chromanoanagenesis in the same patient.
Erythroderma, as Harper-Kirsey comments, is “a severe and potentially life-threatening dermatitis described as an intense, widespread erythema that tends to affect more than 90% of the body surface area, with a variable degree of exfoliative desquamation of the skin and can be a manifestation of a wide range of skin and systemic diseases, including infections, malignancies, and drug hypersensitivity reactions” [11,12,13]. In this context of possibilities, Erythroderma is described as a dermatologic-type paraneoplastic syndrome frequently present in lymphoproliferative neoplasms [14].
Paraneoplastic syndromes are indirect manifestations of cancer not attributable to the direct invasion of the tumor or to compression effects that it may produce and many are expressed before the diagnosis of cancer, so they can serve as sentinel signs. They can be endocrinological (inappropriate secretion of antidiuretic hormone, hypercalcemia, Cushing's syndrome, hypoglycemia), neurological (limbic encephalitis, cerebellar degeneration, myasthenia gravis), dermatological and rheumatological (dermatomyositis, acanthosis nigricans, erythroderma, hypertrophic osteoarthropathy) [14].
In this case, there were strong suspicions regarding a T-cell lymphoproliferative syndrome as a background diagnosis, and our patient had a history of approximately 7 years of evolution with the diagnosis Erythroderma desquamativa, having excluded drug allergies or other common causes. On the other hand, lymphoproliferative disorders in some cases may have incubation times longer than 5 years [15].
Although a definitive diagnosis was not possible on this occasion, the presence of these genetic changes and the concomitant presence of Erythroderma in this patient, reinforce the diagnostic hypothesis of a lymphoproliferative neoplastic picture. Chromoanagenesis can be the cause or the overt consequence of an ongoing massive disorganization process, and can tell us about the ominous character of the condition and its prognosis, providing the treating physicians with important information about the patient's status in extremely complex and difficult to manage cases. When the specific details of the diagnostic and treatment algorithms of chromogenic anagenesis become available, they will provide additional tools for the timely approach to these clinical situations.
New techniques such as OGM (Optical Genome Mapping) will support the finding of these changes for a better approach to these phenomena [16].
The assessment of the degree of chromoanagenesis could perhaps be used in the future as an indicator of the degree of progressive disorganization of the system or in cases of cancer patients and that it would be necessary to reverse to recover homeostasis, as when other parameters are measured in degrees of multisystemic disorganization that allow us to make predictions in specific cases [17].
Conclusions
The use of whole genome analysis techniques are useful tools to clarify the diagnosis of critically ill patients in whom other data are lacking to allow treating physicians to identify the underlying clinical situation underlying severe processes. These techniques, together with oncogenetic evaluations, can provide key pieces in the diagnostic algorithm and management of these patients.
It is also important to be very attentive to the patient's history, in relation to possible sentinel manifestations of dermatological, rheumatological, neurological and endocrinological type that have not been fully clarified.
References
Holland AJ, Cleveland DW. Chromoanagenesis and cancer: mechanisms and consequences of localized, complex chromosomal rearrangements. Nat Med. 2012 Nov;18(11):1630-8. doi: 10.1038/nm.2988. Epub 2012 Nov 7. PMID: 23135524; PMCID: PMC3616639.
Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, McLaren S, Lin ML, McBride DJ, Varela I, Nik-Zainal S, Leroy C, Jia M, Menzies A, Butler AP, Teague JW, Quail MA, Burton J, Swerdlow H, Carter NP, Morsberger LA, Iacobuzio-Donahue C, Follows GA, Green AR, Flanagan AM, Stratton MR, Futreal PA, Campbell PJ. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011 Jan 7;144(1):27-40. doi: 10.1016/j.cell.2010.11.055.
De Pagter M.S., van Roosmalen M.J., Baas A.F., Renkens I., Duran K.J., van Binsbergen E., Tavakoli-Yaraki M., Hochstenbach R., van der Veken L.T., Cuppen E., et al. Chromothripsis in Healthy Individuals Affects Multiple Protein-Coding Genes and Can Result in Severe Congenital Abnormalities in Offspring. Am. J. Hum. Genet. 2015;96:651–656. doi: 10.1016/j.ajhg.2015.02.005.
Devereux TR, Risinger JI, Barrett JC. Mutations and altered expression of the human cancer genes: what they tell us about causes. IARC Sci Publ. 1999;(146):19-42.
Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, McLaren S, Lin ML, McBride DJ, Varela I, Nik-Zainal S, Leroy C, Jia M, Menzies A, Butler AP, Teague JW, Quail MA, Burton J, Swerdlow H, Carter NP, Morsberger LA, Iacobuzio-Donahue C, Follows GA, Green AR, Flanagan AM, Stratton MR, Futreal PA, Campbell PJ. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011 Jan 7;144(1):27-40. doi: 10.1016/j.cell.2010.11.055.
Pellestor, F. Chromoanagenesis: cataclysms behind complex chromosomal rearrangements. Mol Cytogenet 12, 6 (2019). https://doi.org/10.1186/s13039-019-0415-7
Rasnic R, Linial M. Chromoanagenesis Landscape in 10,000 TCGA Patients. Cancers (Basel). 2021 Aug 20;13(16):4197. doi: 10.3390/cancers13164197.
Suzuki E, Shima H, Toki M, Hanew K, Matsubara K, Kurahashi H, et al. Complex X-chromosomal rearrangements in two women with ovarian dysfunction: implications of chromothripsis/chromoanasynthesis-dependant and-independent origins of complex genomic alterations. Cytogenet Genom Res. 2016;150:86–92.
Fukami M, Kurahashi H. Clinical consequences of chromothipsis and other catastrophic cellular events. Methods Mol Biol. 2018;1769:21–33.
Wang K, Wang Y, Collins CC. Chromoplexy; a new paradigm in genome remodelling and evolution. Asian J Androl. 2013;15:711–2.
Harper-Kirksey K. Erythroderma. Life-Threatening Rashes. 2018 Sep 12:265–77. doi: 10.1007/978-3-319-75623-3_19.
Khaled A, Sellami A, Fazaa B, Kharfi M, Zeglaoui F, Kamoun MR. Acquired erythroderma in adults: a clinical and prognostic study. J Eur Acad Dermatol Venereol. 2010;24:781–8.
Usatine RP, Smith MA, Chumley HS, Mayeaux EJ Jr. Erythroderma. In: Usatine RP, Smith MA, Chumley HS, Mayeaux Jr EJ, editors. The color atlas of family medicine. 2nd ed. New York: McGraw-Hill; 2013.
Pelosof LC, Gerber DE. Paraneoplastic syndromes: an approach to diagnosis and treatment. Mayo Clin Proc. 2010 Sep;85(9):838-54. doi: 10.4065/mcp.2010.0099. Erratum in: Mayo Clin Proc. 2011 Apr;86(4):364.
Kaaks R, Sookthai D, Łuczyńska A, Oakes CC, Becker S, Johnson T, Johansson A, Melin B, Sjöberg K, Trichopoulos D, Trichopoulou A, Lagiou P, Mattiello A, Tumino R, Masala G, Agnoli C, Boeing H, Aleksandrova K, Brennan P, Franceschi S, Roulland S, Casabonne D, de Sanjose S, Sánchez MJ, Huerta JM, Ardanaz E, Sala N, Overvad K, Tjønneland A, Halkjær J, Weiderpass E, Bueno-de-Mesquita HB, Vermeulen R, Peeters PH, Vineis P, Kelly RS, Khaw KT, Travis RC, Key TJ, Riboli E, Nieters A. Lag times between lymphoproliferative disorder and clinical diagnosis of chronic lymphocytic leukemia: a prospective analysis using plasma soluble CD23. Cancer Epidemiol Biomarkers Prev. 2015 Mar;24(3):538-45. doi: 10.1158/1055-9965.EPI-14-1107. Epub 2014 Dec 26.
Nilius-Eliliwi V, Gerding WM, Schroers R, Nguyen HP, Vangala DB. Optical Genome Mapping for Cytogenetic Diagnostics in AML. Cancers (Basel). 2023 Mar 9;15(6):1684. doi: 10.3390/cancers15061684.
Hernández Guillamet G, Morancho Pallaruelo AN, Miró Mezquita L, Miralles R, Mas MÀ, Ulldemolins Papaseit MJ, Estrada Cuxart O, López Seguí F. Machine Learning Model for Predicting Mortality Risk in Patients With Complex Chronic Conditions: Retrospective Analysis. Online J Public Health Inform. 2023 Dec 28;15:e52782. doi: 10.2196/52782.
