
Introduction
Apert syndrome (AS) is a rare and complex disorder that is included in the group of acrocephalosyndactylies, which are characterized by malformations in the skull, face, hands, and feet [1]. Most cases result from de novo mutations in individuals without previous family history, although it can also be inherited in an autosomal dominant manner [2]. These mutations affect the FGFR2 gene, which encodes fibroblast growth factor receptor 2 [3]. Specifically, the mutation in the FGFR2 gene involves a cytosine to guanine substitution at position 755, resulting in a change from serine to tryptophan in the resulting protein. This alteration affects the development of the skeletal system, causing premature fusion of the skull bones and inadequate limb formation during the embryonic stage [3,4].
Among the most frequent manifestations that accompany this pathology, we can find craniosynostosis, hypoplasia of the mid-face, and syndactyly of hands and feet [5]. Cardiovascular, neurological, and genitourinary anomalies may also be present [6]. On the other hand, prenatal diagnosis is complex due to the overlap with other craniosynostosis syndromes such as Crouzon, Pfeiffer, Muenke, and Saethre-Chotzen syndromes [6,7]. Additionally, ultrasound studies may suggest craniofacial deformities, but the definitive diagnosis is confirmed postnatally through molecular analysis that identifies mutations in the FGFR2 gene [6]. The aim of this article is to describe and analyze the genetic and molecular characterization of a patient diagnosed with Apert Syndrome. We will address her clinical presentation, molecular diagnosis, and the comprehensive approach of multidisciplinary treatment applied.
Methodology
Prior informed consent was obtained and a detailed medical history was taken from the patient's mother in order to develop the medical record, with special emphasis on investigating the socio-economic conditions during pregnancy, as well as possible exposures to environmental factors that may have had a negative impact on the pregnancy. Additionally, a thorough physical examination was conducted, documenting each of the patient's anatomical-pathological details. Subsequently, blood samples were collected for paraclinical and molecular tests, along with imaging studies, which were essential to confirm the diagnosis of Apert syndrome.
Results
31-year-old pregnant female, with obstetric formula G2P0A1, who during previous prenatal check-ups presents normal paraclinical exams and clinical findings, in adequate socioeconomic conditions and without harmful exposures to environmental factors. However, during the detailed anatomical ultrasound performed at 30.5 weeks, left fetal ventriculomegaly is identified, so she is referred to the perinatologist gynecologist. He decides to terminate the pregnancy by scheduled cesarean section given the previously described findings and orders postnatal studies for ventriculomegaly. At 39.2 weeks of gestation, a newborn female weighing 3,310 grams and measuring 52 cm was born, with adequate neonatal adaptation.
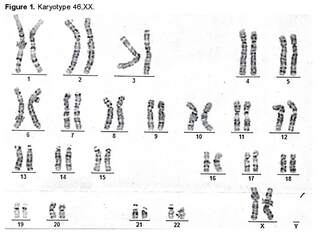
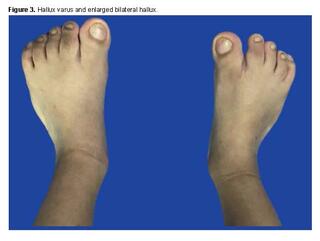
During the first month of life, the following phenotypic findings were identified: right strabismus, right proptosis, hyperostosis of the right coronal suture, craniofacial deformity, right frontal flattening, and increased size of the bilateral hallux [1]. Cytogenetic and diagnostic imaging studies were requested. A G-banding karyotype was performed, in which an analysis of 25 metaphases at a resolution of 400 - 500 bands was conducted, revealing a chromosomal constitution of 46,XX without evident structural alterations (Figure 1).
Additionally, due to cranial asymmetry caused by right fronto-orbital deformity, radiological confirmation of craniosynostosis was performed, leading to the recommendation for surgical correction.
The surgical intervention was performed when the patient was 5 months old, in which findings of craniofacial deformity compromising the right orbit due to decreased horizontal diameter, right frontal depression compensated by left bulging, and hyperostosis of the right coronal suture with pathological fusion consistent with anterior plagiocephaly craniosynostosis were found. The correction and remodeling of the orbit were carried out without complications, with subsequent management in the Pediatric Intensive Care Unit until discharge due to presenting an adequate clinical evolution.
In the presence of suspicion of Pfeiffer syndrome, a molecular panel of 38 genes for craniosynostosis is requested (Table 1), in which a pathogenic heterozygous variant is identified in the FGFR2 gene (cytosine to guanine change at position 755 of cDNA, in exon 7, producing a missense change of serine to tryptophan at amino acid 252). An uncertain significance variant is also found in the same gene (cytosine to thymine change at position 532 of cDNA, in exon 5, which at the protein level produces a missense change of arginine to cysteine at amino acid 178). <<>> With the support of various specialties, a medical board meeting is held to address the internal tibial torsion and instability in the gait of the patient.

The decision was made to use Dennis Brown type orthoses with external rotation at 45 degrees and anti-rotational bands on both feet to align the lower limbs and improve the walking pattern. The patient was readmitted at 2 years of age for a recurrence of craniosynostosis. On the same day, a second surgery and bifrontal decompressive craniectomy were performed, revealing deformities due to fusion of the coronal suture, left frontal bulging, and retreat of the upper orbital rim. The procedure was successful, with transfer to the Pediatric Intermediate Care Unit. The patient developed postoperative anemia, requiring a transfusion of 180cc of red blood cells. She was discharged on the fifth day with a favorable evolution. In the follow-ups after the second surgical intervention, the patient developed a surgical site infection, which was managed with Trimethoprim/Sulfamethoxazole, achieving an adequate response and resolution of the condition.
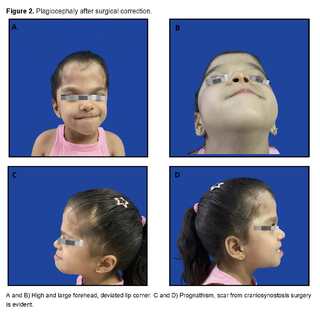
Apart from this complication, there are no other problems derived from the surgery. Figures 2 and 3 show images of the patient, taken at 5 years of age.
Discussion
Apert syndrome, also known as acrocephalosyndactylia type 1, is an autosomal dominant genetic disorder characterized by premature cranial synostosis, facial anomalies, and symmetrical syndactyly in hands and feet. This condition results from point mutations in the FGFR2 gene (fibroblast growth factor receptor 2), located on chromosome 10q26.13. The most prevalent associated mutations are S252W and P253R, which alter the normal development of bones and connective tissues, resulting in the distinctive physical characteristics of this disorder [8,9]. This genetic disorder has an estimated incidence ranging from 1 in 65,000 to 200,000 births, affecting both sexes equally.
It has been observed that the incidence increases with advanced paternal age, suggesting a possible selective advantage in male spermatogonial cells. Although the condition presents complete penetrance, its expressivity is variable, resulting in a range of manifestations that can vary from phenotypically unaffected individuals to cases with severe deformities, even within the same family [10,11,12]. In the clinical presentation of the patient, various phenotypic manifestations characteristic of this syndrome were observed, which have been reported in the literature such as craniosynostosis with craniofacial deformities like hyperostosis of the coronal suture and alterations in the right orbit. Premature closure of the cranial sutures is a distinctive feature of the syndrome, causing an alteration in the shape of the skull [13].
This synostosis mainly occurs in the coronal sutures and is almost universally present in patients with type I acrocephalosyndactyly. However, there have also been documented cases where individuals present other manifestations typical of the syndrome without the characteristic craniofacial morphological alterations [14,15]. In relation to facial development, hypoplasia of the middle facial third contributes to a flattened face, which promotes the development of shallow orbits and eyelid fissures [13]. Underdevelopment of the maxillary structures leads to a class 3 malocclusion, a universally observed feature, and mandibular prognathism. These alterations are associated with exorbitism, strabismus, hypertelorism, low-set ear lobes, and a small-sized nasal structure with a bulbous tip [14,15].
However, the patient only presented, among other anomalies, right strabismus, right proptosis, and increased size of bilateral hallux. Likewise, difficulty with sucking, eating, and swallowing is common in Apert syndrome, partly attributed to a high-arched or cleft palate and an inadequately sized pharynx. Narrowing of the nasal turbinates can cause breathing difficulty. Hearing loss, frequent in these patients, is due to abnormalities in the middle ear, ossicles, and stenosis or atresia of the external auditory canal [14,15]. Syndactyly, another prominent feature of the syndrome, presents symmetrically in the extremities, however, was not observed in this patient. In particular, the hand shows fusion of the second, third, fourth, and fifth fingers, which share bony and soft tissue components, as well as a common nail bed.
The first finger, however, remains mobile and without morphological alterations [14,15]. Finally, anomalies in the central nervous system can be observed, such as malformations of the corpus callosum and limbic system, ventriculomegaly, defects in the gyri, and alterations in white and gray matter. Although intellectual alterations are rare, no significant behavioral problems have been reported. In the cardiovascular system, atrial or ventricular communication, permeable foramen ovale, and aortic override may occur. In the genitourinary system, hydronephrosis and cryptorchidism may be found [14,15]. The approach to craniosynostosis in Apert Syndrome includes a complete clinical history, focusing on family history, exposure to teratogens during pregnancy, and possible reasons for intrauterine head compression (oligohydramnios, abnormal presentation, multiple pregnancy).
Furthermore, a complete physical examination must be performed to confirm craniosynostosis, assessing the shape of the skull from all directions, measuring head circumference, and cephalic index. Postnatally, computed tomography with three-dimensional reconstruction is the standard diagnostic modality, as X-rays and magnetic resonance imaging are less sensitive in determining suture patency [16]. The diagnosis of Apert syndrome is established by identifying classical or suggestive clinical features, along with the detection of a pathogenic heterozygous variant in the FGFR2 gene through molecular testing [17]. Once the phenotypic findings of Apert syndrome are confirmed, mono- or multigene genetic tests can be performed. It is recommended, at a minimum, to perform tests for the pmutation.
Pro250Arg in the FGFR2 gene and in exons 7 and 8, especially in patients with coronal synostosis and multisuture [18]. Monogenic tests detect intragenic deletions or insertions, and missense, nonsense, and splice site variants. However, they do not detect exon deletions or duplications or the complete gene. On the other hand, multigene panels, which include FGFR2 and other genes of interest, have a higher likelihood of identifying the genetic cause of the patient's condition. These panels vary depending on the laboratory and, in some cases, include customized packages [17]. Prenatal diagnosis is difficult before the third trimester, although craniofacial deformities often become more evident after birth. However, findings such as heart and central nervous system diseases (e.g., mild ventriculomegaly or agenesis of the corpus callosum) may be indicative of SA [19].
Inclusive, atypical presentations have been described, including diaphragmatic hernia [20]. Magnetic resonance imaging and ultrasound are useful tools in these cases, and specific findings on prenatal ultrasound may include an elevated skull, agenesis of the corpus callosum, ventriculomegaly, hypoplasia of the midface, hypertelorism, and shallow orbits [21]. The treatment of Apert syndrome requires a multidisciplinary approach. It is essential to undergo surgery in the first year of life to prevent complete closure of the coronal suture and protect brain development. This early intervention offers optimal surgical outcomes and prevents complications such as physical deformities, insufficient cranial volume, and elevated intracranial pressure [22]. Based on the above, the suggested surgical treatment is described according to the stage of life in which the patient is: Treatment by Stages [23]:
Birth at 2 years: Two-stage cranial vault expansion surgery. First, early anterior decompression with a craniotomy as the main procedure, followed by posterior cranial vault expansion. This is done to prevent increased intracranial pressure. From 2 to 12 years: Surgery on the midfacial third, including facial bipartition and distraction osteogenesis aimed at dividing the frontal bone from the supraorbital rim to release the orbits and the midface from the skull base. Additionally, it has an aesthetic purpose as the procedure normalizes the position of the zygomatic bone, nose, and maxilla in relation to the mandible.
From 12 years of age to adulthood: Le Fort II or III surgery and mandibular osteotomy to correct class III malocclusion and, sometimes, open bite, which are performed when the patient turns 17 or 18 years old. Upon reviewing various bibliographic sources, controversy is observed regarding the timing and technique of the first surgical intervention. Some authors suggest performing it before 6 months, while others recommend postponing it due to the possible deterioration of postoperative cranial growth [22,23]. However, there is consensus that the prognosis depends on the severity of the cranial malformation and the possibility of early surgical interventions [24]. Dental treatment is divided into two phases. The first phase, initiated at 8 or 9 years old, corrects anterior crowding and uses fixed or removable mandibular expansion devices. A proper dental assessment at 6 years old can decrease dental impaction and crowding [23]. Treatment of limb malformations.
The correction of syndactyly depends on the degree of tissue involvement and may include orthopedic and plastic surgeries to release soft tissues and improve fine motor skills. For foot deformities, procedures such as syndactyly separation, and the use of casts, splints, and orthoses for adduction or supination deformities can be performed. Early treatment is crucial to prevent gait problems and improve the patient's quality of life [24,25]. Additional Treatments Additional treatment includes repairing cleft palate, surgical correction of strabismus, temporary management of the airway (such as tracheostomy), and treatment of sleep apnea. Furthermore, feeding and language therapies are essential for the proper development of the child [25]. Future Outlook
In the molecular panel study conducted on the patient, a variant of uncertain significance (VUS) was identified. Genetic variants are classified as pathogenic, likely pathogenic, of uncertain significance, likely benign or benign. VUS indicate that current data is insufficient or contradictory to determine their impact on the disease, complicating their interpretation due to the lack of information about their relationship with pathological phenotypes and their frequency in the population. The identification of VUS presents multiple challenges, such as clinical interpretation and genetic counseling. [26, 27, 28]
To manage these variants, a multidisciplinary evaluation and the use of population frequency databases, clinical significance databases, and bibliographic databases, along with bioinformatic tools such as ClinVar, ExAC, LOVD, MutationTaster, among others, are required to obtain a more accurate interpretation of their significance [29]. For better understanding, functional studies and family segregation can provide additional useful information for a possible reclassification of these variants [30]. It is essential to focus on variants predicted as pathogenic, likely pathogenic, or of uncertain significance (VUS). The analysis process includes several steps: first, consulting databases of healthy populations and pathogenic mutations; second, confirming the variant and performing a segregation analysis; and third, conducting an in silico analysis.
If after these analyses the variant continues to be a strong candidate to be a deleterious mutation responsible for the disease, additional functional studies can be considered, among which are tests on platforms and/or studies in knockout mice with the variant to determine their biological functions and involvement in the phenotype [31]. The interpretation of variants of uncertain significance improves with the accumulation of genetic data and the advancement of biological data processing technologies. It is essential to keep databases updated and to use more precise analysis techniques to characterize these variants more effectively [32]. Including these variants in laboratory reports with recommendations for follow-up and periodic reassessment is essential for accurate diagnosis and clinical management [33].
Furthermore, some variants that currently have an uncertain or unknown significance could acquire a pathological relevance in the future, highlighting the importance of continuous reevaluation [34].
Conclusions
Apert syndrome (AS) is a rare genetic disorder, which makes early diagnosis have a significant impact on prognosis and quality of life of patients. This syndrome often presents respiratory and central nervous system complications. Therefore, an early multidisciplinary approach can improve the life expectancy of patients, as their prognosis depends on clinical severity and timely treatment. The detection of variants of uncertain significance (VUS) presents diagnostic challenges that require specialized evaluation and the use of appropriate tools.
The constant updating of databases and regular monitoring are essential, since some variants may acquire pathological relevance over time [1].