
INTRODUCTION
Osteogenesis imperfecta (OI), known as a disorder characterized by bone weakness and high risk of fractures, of genetic origin, is known as brittle bone disease [1]. It has been found that this pathology generates a high burden of morbidity and mortality, associated with the presence of pain and functional limitations for those who have it [1]. The fragility present in the bones has been described by structural variants of the Collagen 1 genes (COL1A1 and COL1A2), among others [1,2]. According to population studies, an estimated prevalence between 1/15,000 and 1/20,000 cases in live births is known, being classified as a orphan disease [3,4]. Throughout history, different types of classifications have been presented since the initial classification in 1979 by Sillence, to the clinical classification of 2014, and through classifications based on findings of genetic variants [1,2,16].
According to these classifications, OI can have its etiology in different genetic variants and not only alterations of the COL1A1 collagen gene, however, it is the gene with the greatest implication in this pathology. Any alteration of the long arm of chromosomes 7 and 17 can cause a alteration in the amount of pro-collagen synthesized, with approximately up to half of type I collagen being affected in quantity or quality. So far, more than 150 variants related to this gene have been reported, which can result in OI [6,7]. The main characteristic is the presence of numerous fractures or bone deformities, due to the great fragility, however, there is a great phenotypic heterogeneity [8]. There are severe presentations related to congenital conditions, which include fractures in utero and during childbirth, with serious complications at the brain and lung levels, fatal.
Other cases of mild to moderate presentation, with fewer fractures, more bone deformity, and some other complications. That is why today the clinical classification is based on severity and genetics based on molecular levels [5,7,8]. They also present characteristic facial features, with a wide and prominent forehead with large temporal and parietal bones, blue sclerae due to intraocular pigments, muscular atrophy due to the number of fractures, thin skin, and capillary fragility. 80-90% of patients have scoliosis and basilar impression can be found in up to 2% of patients in the cervical spine [1,5,6]. Osteogenesis Imperfecta type I is described as the most common presentation, affecting up to two-thirds of the diagnosed population.
It is generally associated with milder severity, with some of the clinical characteristics such as bone fragility, blue sclerae, conductive hearing loss, and minimal bone deformity [1,8]. Timely diagnosis of the disease allows for the initiation of therapeutic alternatives that help reduce the burden of the disease, such as the number of fractures, pain management, and multidisciplinary treatment with growth hormone, anti-resorptive bone therapy, anabolic therapies, or even anti-TGFβ antibodies [3]. Numerous research studies on new treatment strategies have been developed with the aim of inhibiting excessive bone resorption or increasing bone formation [8-10].
Among the most promising treatments are denosumab, a monoclonal antibody that acts against the activator of the NF-kappa B ligand receptor, a key cytokine of osteoclasts; and monoclonal antibodies against anti-sclerostin proteins, and dickkopf-1 endogenous inhibitor of bone formation, however these are still in the study phases [11,12]. Additionally, there are studies that report autologous bone marrow transplantation, however, without conclusive results yet. Therefore, a multidisciplinary approach with an emphasis on physical rehabilitation is still being considered to improve motor activity, surgery if needed to correct deformities. Additionally, pharmacological treatment and genetic counseling [11,12] are recommended.
The objective of this clinical case is to raise awareness among healthcare personnel about the importance of timely recognition of this pathology and the impact achieved by making an early and accurate diagnosis of the condition. Despite the phenotypic variability found in this disease, it is of great importance to carry out genetic studies and counseling as they can present as de novo variants, which may appear for the first time in a family, not inherited, secondary to changes in a germ cell (ovum or sperm) or in the zygote; and their identification will allow to start targeted therapeutic alternatives, in order to reduce the burden of morbidity and mortality of the patients who suffer from it. And provide follow-up guidelines, prognosis, moving closer to personalized and precision medicine.
CLINICAL CASE
Eight-year-old female patient with a clinical history of fractures secondary to mild traumas since the age of 4 years old. The first fracture was in the left foot metatarsal with the mechanism of trauma being a jump from her own height, the second fracture occurred at 6 years old in the right hand finger after falling off a bike, and at 7 years old she fractured her right foot after falling from her own height. Other diagnoses include bilateral otosclerosis requiring hearing aids and early puberty. She is the child of non-consanguineous parents, with no known family history of genetic connective tissue diseases.
During the physical examination, the patient presented with facial dysmorphia associated with a wide, prominent forehead, slightly blue-tinted sclera, irregular teeth with dentin wear, bilateral hearing aids, described scar from fractures, breast bud present, dark and sparse hair without signs of estrogenization, ligamentous hyperlaxity, normal behavior and cognition, with no other alterations on physical examination.
In terms of paraclinical approach, the patient had a report of urinalysis showing urinary sediment calcium +++ and phosphorus of 5.87 mg/dL (elevated), and calcium of 10.56 mg/dL (elevated), with the rest of the paraclinical tests showing normal bone metabolism.
He had imaging studies of total spinal column x-ray with mild dextroconvex lumbar scoliosis, and anterolisthesis of L5 over S1, simple skull x-ray without fractures or alterations, and long bone x-rays - complete series showing previously described fractures, with renal and urinary tract ultrasound revealing mild bilateral ectasia and a 15% residual in bladder emptying.
Due to clinical history and physical examination with suspicion of Collagen Tissue Disease - Collagenopathy, a molecular study (sequencing + CNVs through NGS) was requested for genes related to this medical condition.
RESULTS
Molecular study sequencing + CNVs through NGS for genes related to collagenopathies in which a heterozygous pathogenic variant is detected in the COL1A1 gene (See Table 1).

This variant generates the change of a guanine to an adenine at position 3,652 of the cDNA, in exon 48 of the gene (c.3652G>A), and in the protein it produces the erroneous change of an alanine to a threonine at amino acid 1,218 (p.Ala1218Thr), an evolutionarily conserved amino acid. Another nucleotide change has been previously documented at this position that has been classified as deleterious (chr17:48,264,163 C => G (Ala1218Pro)), which supports the clinical suspicion of hereditary collagenopathy.
Deleterious variants in the COL1A1 gene are associated with autosomal dominant type I osteogenesis imperfecta (see details in supplementary table).
DISCUSSION
In Osteogenesis Imperfecta, there is a variant in the COL1A1 or COL1A2 genes in up to 90% of cases. This affects type I collagen fibers, which play a role in bone strength [1,15].
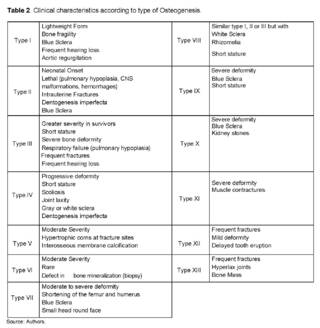
The Sillence classification [16], which was the first classification in 1976, did not take into account variations in genes and only had some classifications based on clinical criteria. Nowadays, a classification of the disease is carried out both clinically and genetically. In Table 2, the clinical characteristics specific to each type of osteogenesis are observed [11,12,18].
The case described corresponds to a patient with clinical and imaging suspicion of collagenopathy, with molecular confirmation of mild phenotype type I osteogenesis imperfecta, with clinical findings that allow the establishment of said genotype-phenotype correlation, frequent fractures, hearing loss, and blue sclerae. Although otosclerosis is not one of the most frequent findings in the population with osteogenesis imperfecta, there is a study reporting a family with several members affected by both disorders [14].
According to this study, otosclerosis is present in 12% of Caucasian patients, with a higher prevalence in women, and there are two cases of a family with osteogenesis imperfecta and otosclerosis [1]. However, in our patient, no relatives with other auditory or genetic disorders like osteogenesis imperfecta were found. For the reported inheritance in osteogenesis imperfecta type I, there are findings of autosomal dominant inheritance, mainly in the COL1A1 or COL1A2 genes, but there are also some reported variants in genes CRTAP, PPIB, SP7 [9]. In our patient, molecular study evaluated 68 genes related to collagen disorders (ACTA2, ADAMTS2, ALDH18A1, ATP6V0A2, ATP7A, B3GALT6, B3GAT3, B4GALT7, BGN, C1R, CBS, CHST14, COL11A1, COL12A1, COL1A1, COL1A2, COL2A1, COL3A1, COL4A1, COL5A1, COL5A2, COL5A3, COL6A1, COL6A2, COL6A3, among others) where only this variant was found.
In this case, a change from guanine to adenine was evidenced in the COL1A1 gene at position 3652 of the cDNA, in exon 48 of the gene (c. 3652G>A), which at the protein level produces a change from alanine to threonine at amino acid 1218 (p. Ala1218Thr). As shown in Figure 1 [21], different systems are used to observe the changes presented in the gene and the possible products of these variants. Additionally, a search for this variant was conducted in artificial intelligence assistants. According to the first genetic variant assistant from GenAI, VarChat [22]: the genomic variant c. 3652G>A is a single nucleotide substitution that occurs in the COL1A1 gene, where guanine (G) is replaced by adenine (A) at position 3652 of the DNA nucleotide.

The change results in the alteration of the codon for alanine (Ala) at position 1218 in the protein sequence to a codon for threonine (Thr), referred to as p. Ala1218Thr. The variant is cataloged in public databases with the reference SNP ID rs72656337 (See Figures 1 and 2). Genetic function: the COL1A1 gene encodes the pro-alpha1(I) chain of collagen type I, which is an important structural component of the extracellular matrix in various connective tissues throughout the body. Type I collagen is a fibrillar collagen, formed by the association of two pro-alpha1(I) chains and one pro-alpha2(I) chain, creating a triple helix structure. This protein plays a key role in the strength and elasticity of connective tissues, including bones, skin, and tendons.

Clinical significance
The variant rs72656337 has been reported in ClinVar with a total of 1 pathogenic presentation.
The substitution at position 1218 is located within one of the collagen triple helix domains, which are crucial for the formation and stability of collagen fibrils.
Substitution of the non-polar alanine residue with the polar threonine residue may alter the helical structure, which could lead to changes in the formation and function of the fibrils. These structural changes may have important implications for the mechanical properties of connective tissues.
Pathogenic variants in the COL1A1 gene are known to be associated with various connective tissue disorders, including osteogenesis imperfecta (OI), a group of genetic disorders characterized by bone fragility, low bone mass, and susceptibility to fractures. The clinical presentation of OI can vary widely, from mild to severe, depending on the specific nature of the collagen defect.
In conclusion, the c. 3652G>A, p.Ala1218Thr variant in the COL1A1 gene is a nonsense change that has been associated with pathogenic outcomes in the context of connective tissue disorders. The clinical importance reported in ClinVar supports the potential impact of this variant on protein function and disease manifestation. Treatment should be multidisciplinary and based on the patient's clinical condition. Recent studies have shown the use of Setrusumab as a biological agent in managing bone calcium fixation. However, these studies have only been conducted in adults [19]. There is only one study in children comparing the use of Setrusumab and bisphosphonates, but this study is still in phase 3, with further studies needed on the prevalence of this type of osteogenesis and pharmacogenomics [20].
CONCLUSION
Osteogenesis imperfecta is a rare genetic disease with phenotypic, endotypic, and genotypic variability, which is why undertaking strategies for early identification impacts the natural history of the disease, contributing to the development of preventive, personalized, predictive, precise, participatory medicine in order to be carried out at a population level. The different databases, different high-performance bioinformatics algorithms, individual predictors, and MetaScore, together with knowledge about functionality, biological bases, genomic and molecular annotation data, protein structure and function, functional studies, use of artificial intelligence tools, and in accordance with Richards, et al.
Standards and guidelines for the interpretation of sequence variants [23], by the American College of Medical Genetics and Genomics, Association for Molecular Pathology, ClinGen, classify this variant as likely pathogenic. Using the artificial intelligence genomic platform Mastermind, it is classified as PM2, PM1, PP2, PP1M, PPC, PPC Het with NM_000088.4:c.3652G>A (p.Ala1218Thr) Pathogenic, which establishes a genotype/endotype/phenotype correlation. Through this case, the importance of a diagnosis and genotype-phenotype correlation that allows for targeted actions to be taken, understanding specific pathophysiological mechanisms that allow for the sub-classification of collagenopathies, contributing to the increase in medical-scientific knowledge about disease expression and the trend towards hyper-personalization, is sought to be raised awareness.
ABOUT THE AUTHORS
Jhonatan Alzate-Valencia
Physician, Resident Specialization in Pediatrics, Research Group in Pediatrics (GRINPED), Neurogenetics and metabolic diseases research line.
Lina-Johanna Moreno-Giraldo
Physician, Specialist in Pediatrics, Master and PhD in Medical Genetics, Research Group in Pediatrics (GRINPED), Neurogenetics and Metabolic Diseases Research Line. Professor, Department of Clinical Postgraduate Studies.