
Variantes genómicas asociadas a errores congénitos en el metabolismo de los carbohidratos en el suroccidente colombiano
Genomic variants associated with inborn errors in carbohydrate metabolism in southwestern ColombiaJaime David Viafara Belalcazar1,2,3
 , Jose María Satizabal Soto1,2,3 , Lina Johanna Moreno Giraldo1,2,3
, Jose María Satizabal Soto1,2,3 , Lina Johanna Moreno Giraldo1,2,3 1. Universidad del Valle, Cali, Colombia; 2. Universidad Libre, Cali, Colombia; 3. Research Group on Congenital Metabolic Diseases;
Descargas
Resumen
Introducción: Los Errores Innatos del Metabolismo (EIM) de carbohidratos son el resultado de la interrupción de las vías catabólicas o anabólicas de diferentes carbohidratos, siendo la fructosa, la galactosa y el glucógeno los más comunes y pertenecen a un grupo heterogéneo de trastornos que pueden ser hereditarios o pueden ocurrir como resultado de variantes genéticas espontáneas. En los EIM, están las vías de los carbohidratos alteradas, tanto las vías catabólicas como las anabólicas, incluyendo el metabolismo de la fructosa, galactosa y glucógeno, dando lugar a un grupo heterogéneo de trastornos. Sin embargo, los datos sobre estas patologías en Colombia son escasos. Nuestro estudio tuvo como objetivo identificar variantes genómicas asociadas a EIM por carbohidratos en población del suroccidente colombiano carente de diagnóstico clínico. Resultados: Realizamos un estudio transversal, secuenciando 320 exomas y clasificando las variantes de acuerdo a guías estándar. Identificamos 286 variantes, incluyendo 206 nuevas y 73 variantes benignas o probablemente benignas. En particular, seis variantes en genes como GALT, GAK1, ALDOB, GAA y SLC2A1 tenían un significado incierto, y una variante patogénica en el gen GALT se asoció con la galactosemia clásica. Curiosamente, el 18% eran intrónicas, el 5% con sentido erroneo, el 10% sinónimas y el 67% variantes no notificadas. Conclusiones: Estos hallazgos ponen de relieve la necesidad de programas de diagnóstico precoz para aplicar tratamientos específicos, incluido el manejo transdisciplinar para minimizar la morbilidad y la mortalidad. El consejo genético y la educación sobre el riesgo son cruciales, facilitando enfoques de medicina anticipatoria y preventiva, avanzando así en la medicina de precisión.
INTRODUCTION
Los Errores Innatos del Metabolismo (EIM) de los hidratos de carbono o también denominados Trastornos Metabólicos Hereditarios según la Clasificación Internacional de Trastornos Metabólicos Innatos (ICIMD) representan un grupo heterogéneo de trastornos genéticos que afectan a la capacidad del organismo para procesar y utilizar adecuadamente los hidratos de carbono, como los trastornos del metabolismo de la galactosa y la fructosa, el metabolismo del glucógeno y la glucólisis; el metabolismo de las pentosas, el transporte transmembrana de hexosas y la absorción. [1,2]. Los EIM de carbohidratos son el resultado de la alteración de las vías catabólicas o anabólicas de diferentes carbohidratos, siendo los más comunes la glucosa, la fructosa, la galactosa y el glucógeno, con pronósticos para los pacientes afectados que varían desde benignos hasta potencialmente mortales [3]. Asimismo, la literatura existente ha documentado una serie de EIM de carbohidratos de gran interés, como la galactosemia, la intolerancia a la fructosa y la deficiencia de glucosa-6-fosfato deshidrogenasa (G6PDH), entre otras [4].
Estos trastornos son el resultado de variantes en genes clave implicados en las vías metabólicas de los carbohidratos, que afectan la síntesis o degradación de estos compuestos, con una variabilidad genética diferenciada según la población, incluyendo el suroeste de Colombia, que puede dar lugar a perfiles específicos de variantes genómicas asociadas a estos errores congénitos [5]. Estas patologías, aunque raras a nivel individual, son colectivamente comunes, con una incidencia mundial estimada de 1 por cada 1.900 nacimientos [6].
La edad de aparición puede variar desde la infancia hasta la adolescencia e incluso la edad adulta, aunque las formas más graves tienden a manifestarse durante el periodo neonatal y se asocian con una mortalidad significativa [7]. En Colombia, el diagnóstico de laboratorio de estos trastornos metabólicos no es una práctica común en los servicios de salud; sin embargo, gracias al esfuerzo conjunto de los prestadores de servicios de salud y los profesionales de la salud, en los últimos años se han realizado importantes esfuerzos para intensificar la difusión y el conocimiento de estos trastornos [8].
De igual forma, en Colombia se están creando programas de tamizaje neonatal bajo la ley 1980 de 2019 [9]. Esta ley solo contempla seis patologías, como fenilcetonuria, galactosemia, fibrosis quística, hiperplasia suprarrenal congénita, deficiencia de biotinidasa y defectos de hemoglobina, dejando por fuera otros EIM por carbohidratos, como la enfermedad de Pompe (EP) [10]. Además, la implementación avanza lentamente, ya que los actuales programas de cribado neonatal de cobertura total sólo buscan activamente el hipotiroidismo congénito como parte de los programas financiados a nivel nacional [11].
La secuenciación del exoma completo (WES) es cada vez más habitual en la práctica clínica, ya que aborda un mayor número de genes y es capaz de identificar variantes puntuales y otros tipos de alteraciones genéticas en el ADN. Esta técnica ha demostrado su utilidad en el diagnóstico de los EIM, y su coste decreciente la está convirtiendo en un procedimiento estándar en los protocolos de diagnóstico de estos trastornos [12]. Los estudios para identificar nuevas variantes genéticas están ampliando el campo de conocimiento sobre los EIM, lo que favorece la detección precoz y el tratamiento oportuno [13]. Sin embargo, las aproximaciones clínicas para el diagnóstico de los EIM son difíciles, especialmente para las variantes más raras de la enfermedad, lo que representa un reto para los clínicos que deben conocer este grupo de afecciones y aprender a identificar las manifestaciones típicas de los diferentes EIM [14]. A su vez, Colombia carece de estudios que caractericen y determinen el significado clínico de las variantes encontradas en estudios genéticos poblacionales, además, no existen estudios previos de frecuencia alélica que permitan estimar el riesgo de ocurrencia de estas enfermedades en el suroccidente colombiano, teniendo otros factores de riesgo como la endogamia asociada a poblaciones aisladas geográficamente, lo que aumenta el riesgo de ocurrencia de estas patologías de herencia autosómica recesiva en su mayoría [15,16].
En el contexto específico del suroccidente colombiano, la prevalencia y diversidad de variantes genómicas asociadas a estos errores congénitos son áreas de estudio poco exploradas. Por lo tanto, el presente trabajo buscó identificar, determinar y caracterizar las variantes genómicas de los genes asociados a EIM por carbohidratos, a través del análisis de los resultados de exomas completos de 320 pacientes del suroccidente colombiano encontrados en la base de datos genómica del Instituto de Genómica de Genética Médica de la ciudad de Cali, quienes no tuvieran diagnóstico clínico y/o paraclínico de este tipo de patologías, con el fin de determinar sus frecuencias alélicas. Esto permitirá ampliar el conocimiento de estas patologías entre la comunidad médica, científica y las autoridades de salud, con el fin de sugerir el establecimiento de programas de tamizaje y diagnóstico temprano, que permitan la implementación de tratamientos oportunos, ya que existen opciones específicas y dirigidas para algunas de estas patologías.
MATERIALES Y MÉTODOS
Se trata de un estudio transversal, observacional y descriptivo en el que se buscaron, identificaron y caracterizaron genómica y bioinformáticamente las variantes genéticas candidatas de la galactosa-1-fosfato uridiltransferasa (GALT), la galactocinasa 1 (GALK1), la aldolasa, fructosa-bisfosfato B (ALDOB), Alfaglucosidasa (GAA), Transportador de glucosa tipo 1 (SLC2A1) y Glucosa-6-fosfato deshidrogenasa (G6PDH), previamente identificadas mediante revisión bibliográfica teniendo en cuenta la frecuencia de los informes, el impacto clínico, la prevalencia y la incidencia; asociadas a galactosemia tipo I y II, intolerancia hereditaria a la fructosa, enfermedad de Pompe, deficiencia del transportador de glucosa I, deficiencia de glucosa 6-fosfato deshidrogenasa en una población del suroccidente colombiano con diferentes patologías y no diagnosticadas clínica y/o paraclínicamente con EIM por carbohidratos.
Revisión bibliográfica
Para la selección de los genes asociados a los EIM de carbohidratos, se realizó una revisión bibliográfica en las bases de datos Scopus, PubMed y LILACS de artículos publicados entre 2010 y febrero de 2023 (idiomas inglés, español y portugués), incluyendo artículos que se refirieran únicamente a errores innatos del metabolismo de carbohidratos, que proporcionaran información sobre prevalencia, características clínicas y/o información sobre letalidad y que fueran realizados en humanos. Dos investigadores realizaron la búsqueda de forma independiente y un tercer investigador corroboró la información. Posteriormente, se registró la información relevante en una hoja de cálculo Excel para ordenar las publicaciones y resumir los datos principales.
Población de estudio Se utilizaron los resultados obtenidos de la secuenciación del exoma completo de 320 pacientes que no presentaron diagnóstico clínico y/o paraclínico de EIM de carbohidratos del suroccidente de Colombia, los cuales fueron suministrados por el Instituto de Genética Médica - GENOMICS (Cali), previo consentimiento firmado de los participantes.
Criterios de selección
Resultados de exoma completo de pacientes que no presentaron diagnóstico clínico y/o paraclínico de EIM de carbohidratos de la base de datos del Instituto de Genética Médica - GENOMICS (Cali).
- Pacientes de las regiones del suroccidente colombiano.
- Todos los pacientes que firmaron un consentimiento
- Asentimiento informado para el uso de los datos.
Secuenciación del exoma
La secuenciación del exoma completo se realizó en los pacientes mediante extracción de sangre y posterior recogida en papel de filtro e inmersión en tampón fosfato. Extracción del ADN, utilizando el paquete DNeasy de Qiagen; se cuantificó y verificó la calidad y cantidad de ADN de cada muestra. A continuación, se realizó la secuenciación masiva de las bibliotecas Nextera TM utilizando la plataforma Illumina con una cobertura 100X. La alineación de secuencias se realizó con el genoma de referencia GRCh38/hg19. Los resultados de la secuenciación se encuentran en archivos de formato de llamada de variantes (VCF) y fueron sometidos a un primer filtro de interpretación y priorización, todo ello con el objetivo de garantizar la veracidad y calidad de los datos genómicos obtenidos.
Identificación de variants
Todos los exomas pertenecientes al big data del Instituto de Genética Médica - GENOMICS (Cali), 320 pacientes en total, se encuentran en formato VCF (Variant Call Format). Para la identificación de las variantes genéticas se utilizó el programa VCard File (VCF) Editor 1.0, el cual permite procesar el formato VCF de los exomas, aplicar diferentes filtros de búsqueda y obtener un archivo final de hoja de cálculo con la información organizada en columnas por cromosoma, posición, nucleótido alterado, registro Clinvar, entre otras características [17].
Análisis bioinformático
El análisis bioinformático de las variantes se realizó utilizando las bases de datos Clinvar, Varsome e Intervar para identificar si habían sido descritas previamente [18-20]. Asimismo, se determinaron las consecuencias asociadas a la enfermedad utilizando herramientas bioinformáticas, Functional Analysis through Hidden Markov Models (FATHMM), Polymorphism Phenotyping v2 (Polyphen-2), Mutation Taster, MutationAssessor, Protein Variation Effect Analyzer (PROVEAN), Sorting Intolerant From Tolerant (SIFT), UMD-Predictor, Human Splicing Finder.
Clasificación de las variantes
Las variantes genéticas se clasificaron de acuerdo con las normas y directrices para la interpretación de variantes de secuencia del American College of Medical Genetics and Genomics y la Association for Molecular Pathology [21].
Cálculo de las frecuencias alélicas
La frecuencia alélica se calculó por simple recuento, dividiendo el número de alelos totales encontrados para cada variante sobre el número total de pacientes con exoma:
Ecuación 1. Frecuencia alélica por recuento simple [22]:
Número de copias del alelo en la población / Número total de copias del gen en la población
Aspectos bioéticos
El comité de ética de la Universidad del Valle (Cali, Colombia) revisó y aprobó el estudio. Los autores declaran no tener conflicto de intereses. Asimismo, en este artículo se mantuvo la confidencialidad de los datos y se obtuvo el consentimiento informado del representante legal del paciente (padre y madre).
RESULTADOS
Revisión bibliográfica
La estrategia de búsqueda arrojó 706 registros, luego de la exclusión de registros duplicados y la aplicación de los criterios de inclusión, se obtuvieron 94 publicaciones, 47 fueron excluidas luego del análisis de texto completo y 47 estudios fueron incluidos en el análisis final, los cuales reportaron con mayor frecuencia datos sobre los genes GALT, GALK1, ALDOB, GAA, SLC2A1 y G6PD, asociados a EIM por carbohidratos; por lo tanto, estos genes fueron elegidos para ser evaluados en la población de estudio.
Variantes genómicas en EIM de Carbohidratos
A partir del análisis genómico y bioinformático de 320 exomas completos de pacientes del suroccidente colombiano, sin diagnóstico clínico y/o paraclínico asociados a errores innatos del metabolismo de carbohidratos, se encontraron 286 variantes relacionadas con los genes ALDOB, G6PD, GAA, GALK1, GALT y SLC2A1; 191 de estas variantes no habían sido reportadas previamente en las bases de datos Clinvar, Intervar y Varsome, 19 de ellas no tienen significado clínico reportado en estas plataformas. Los genes con el mayor número de variantes encontradas fueron GALK1, GAA y G6PD asociados con galactosemia tipo II, enfermedad de Pompe y deficiencia de glucosa 6 fosfato deshidrogenasa respectivamente; los genes con el menor número de variantes reportadas fueron ALDOB y GALT. Entre las variantes de las que se había informado anteriormente, el 29% eran cambios sinónimos, el 16% eran cambios de sentido erróneo y el 55% eran variantes en regiones no codificantes en los genes asociados a la EIM de carbohidratos (Figura 1).
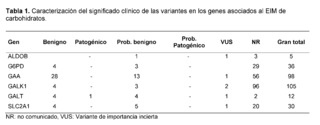
Significado clínico de variantes genómicas en genes asociados a EIM por carbohidratos en el suroccidente colombiano.
Con respecto al significado clínico dado por el ACMG y de acuerdo al software de predicción utilizado en las variantes encontradas, el 24,5% presentó un significado benigno y probablemente benigno, el 3,1% presentó un significado incierto y el 0,3% obtuvo una clasificación de patogénico o probablemente patogénico. Para esta última clasificación, en el gen GAA, el software de modelado y predicción mostró un efecto de patogenicidad en tres variantes (p.Gly576Ser, p.Asp91Asn y p.Thr927Ile); sin embargo, debido a su altísima frecuencia alélica, el 2,5%, 2,2% y 1,6% respectivamente fueron reclasificadas como benignas y probablemente benignas. El 72% restante correspondía a variantes de las que no se ha informado funcionalmente y actualmente se desconoce su importancia clínica.
Frecuencias alélicas encontradas
De acuerdo con las frecuencias alélicas analizadas, la variante en la posición chr17:g.73753661C>T en el gen GALK1 presentó la mayor frecuencia, con un 34,38% en toda la muestra; sin embargo, esta variante no presentó significación clínica reportada en bases de datos como Clinvar y Varsome. De igual forma, la variante c.*22+1116G>C en el gen GALK1 reportó una frecuencia de 32.19%, la cual no ha sido reportada en bases de datos como Clinvar, ni en programas de predicción de regiones intrónicas como Human Splincing Finder, sin embargo, presentó una significación clínica de benigna en otras bases de datos como Varsome.
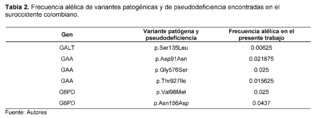
Las variantes que fueron analizadas por el software de predicción y clasificadas como variantes patogénicas, probablemente patogénicas o pseudodeficientes, como se muestra en la Tabla 2, presentaron una frecuencia alélica que osciló entre 0,025 y 0,00625, siendo 0,00625 la frecuencia más baja reportada para la variante p.Ser135Leu del gen GALT.
DISCUSIÓN
En los exomas completos de 320 pacientes con patologías complejas y no diagnosticados clínica y/o paraclínicamente de ningún EIM, se encontraron 286 variantes para los genes elegidos por la literatura: GALT, GALK1, ALDOB, GAA, G6PD y SLC2A1, de las cuales, según los criterios establecidos en las guías emitidas por el ACMG y la Association for Molecular Pathology, se clasificaron en 73 variantes de significado benigno o probablemente benigno, 1 variante de significado patogénico, 6 variantes de significado incierto y 206 variantes que no han sido reportadas previamente en bases de datos clínicas y poblacionales.
Variante patogénica en GALT
La Galactosemia clásica (GC) pertenece a un raro conjunto de trastornos hereditarios del metabolismo de la galactosa, causados por defectos en la enzima galactosa 1-fosfato uridililtransferasa codificada por el gen GALT; como consecuencia del déficit enzimático, los pacientes presentan un amplio espectro de discapacidad física y cognitiva, llegando a ser potencialmente mortal [23]. En cuanto al gen GALT, dentro de los resultados de los 320 exomas completos, la variante c.404C>T (p.Ser135Leu) tuvo un significado clínico de patogenicidad a través del análisis realizado por el software bioinformático, con una frecuencia de 0.00625, la cual se ha descrito que afecta principalmente a pacientes con ascendencia africana y comprende hasta el 90% de las variantes causales de galactosemia en pacientes sudafricanos [24,25]. La variante p.Ser135Leu demostró un efecto patogénico tras el análisis funcional proteico in silico en todos los programas informáticos de predicción empleados, lo que concuerda con los estudios in silico realizados por Kumar S U et al., que emplearon un proceso computacional exhaustivo para la variante sin sentido, p.Ser135Leu, para analizar la conservación de aminoácidos, las predicciones de patogenicidad y la estabilidad de la proteína, utilizando herramientas bioinformáticas, encontrando que la sustitución del aminoácido nativo por una leucina, causa la pérdida de enlaces de hidrógeno en el núcleo de la proteína, lo que altera el plegamiento correcto en la región del bucle y las ocho regiones beta-hoja, concluyendo que p. Ser135Leu en comparación con otras variantes, es la más deletérea entre las variantes prevalentes que alteran el funcionamiento general de la proteína [26].
Esto también es apoyado por los estudios realizados por Coelho AI et al., cuando caracterizaron molecularmente la deficiencia de GALT en Portugal, en cuarenta y dos pacientes galactosémicos donde la variante p.Ser135Leu era la tercera más prevalente en su población (4%), los padres de los pacientes portadores de la variante p.Ser135Leu eran de origen africano [27]. Los estudios realizados por Crespo C, et al., en 2020 en 37 pacientes argentinos con una actividad enzimática de la galactosa-1-fosfato uridililtransferasa inferior al 35% del valor normal, identificaron la variante p.Ser135Leu en un paciente con una actividad enzimática del cero por ciento [28].
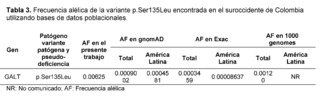
La frecuencia alélica de la variante p.Ser135Leu estaba por encima de las frecuencias latinas comunicadas por ExAC y gnomAD; del mismo modo, esta variante en la población de estudio superaba los datos comunicados en las frecuencias globales (Tabla 3). La prevalencia de variantes africanas en la población del suroccidente colombiano puede estar asociada al alto porcentaje de ancestría africana en el territorio colombiano que puede variar entre 0,0 y 92,1% dependiendo de la población y el grupo étnico de estudio [29].
Variantes de pseudodeficiencia en OAG
Las alteraciones patológicas en el producto proteico del gen GAA se asocian a la enfermedad por almacenamiento de glucógeno tipo II o EP, causada por una disminución de la alfa-glucosidasa ácida, necesaria para la degradación del glucógeno en glucosa, alterando la función del metabolismo celular y lisosomal. [30]. Las variantes del gen GAA representaron el 36% de las variantes encontradas en el presente estudio, donde el 50% de las variantes se encontraban en regiones intrónicas. Con respecto a las variantes encontradas, la variante p.Gly576Ser de pseudodeficiencia común [31] ha sido descrita por Reuser AJJ et all. en heterocigosis en posición cis en 17 pacientes asiáticos con EP clásica infantil con cardiomiopatías menos severas y progresión más lenta de la enfermedad [32]. Asimismo, de Reuser AJJ et al. encontraron otras variantes de pseudodeficiencia; p.Asp91Asn y p.Thr927Ile en pacientes caucásicos afectados con EP de inicio tardío sin cardiomiopatía, con una forma más atenuada con inicio de síntomas antes de los 12 años de edad [32]. Los alelos de pseudodeficiencia p.Asp91Asn y p.Gly576Ser, han sido reportados en una población Americana de ascendencia Asiática, en 38 infantes con screening positivo para EP, siendo de inicio tardío más prevalente [33]. De forma similar, las variantes p.Asp91Asn y p.Gly576Ser fueron descritas en 3 pacientes con sospecha de EP de inicio tardío, presentando otras pruebas bioquímicas alteradas junto con una actividad enzimática muy cercana a los valores mínimos [34] con diversos grados de sarcopenia o síndrome de fragilidad en la edad adulta [35].
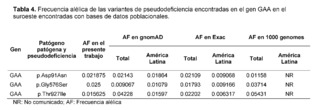
En comparación con bases de datos poblacionales como 1,000 Genomes, ExAC y gnomAD, se encontró que la variante p.Aps91Asn para GAA presentó una frecuencia alélica dentro de los valores de frecuencia global pero muy por encima de lo reportado para Latinoamérica, GnomAD y Exac. La variante Glys576Ser para GAA presentó una frecuencia alélica por encima de la reportada en bases de datos poblacionales que contienen frecuencias génicas de poblaciones latinoamericanas, y esta variante también presentó una prevalencia mucho mayor en la población latina comparada con la frecuencia global de estas bases de datos (Tabla 4).
En Colombia, la EP no está incluida en los programas de tamizaje neonatal, aunque es posible identificarla mediante espectrometría de masas en tándem [33]. Adicional a esto, se han documentado los beneficios del tratamiento antes del inicio de los síntomas clínicos en esta enfermedad progresiva, evitando daños musculares irreversibles [36]. Los resultados de variabilidad y frecuencia alélica del gen GAA son la base para advertir a las autoridades sanitarias competentes sobre la existencia de estas variantes en la población del suroccidente colombiano, reconociendo la importancia de incluir esta patología dentro de los programas de tamizaje neonatal.
Variantes de pseudodeficiencia en G6PDH
La deficiencia de G6PDH es considerada la anormalidad genética más común que se hereda como un rasgo polimórfico ligado al cromosoma X, con un estimado de 400 millones de personas afectadas y su distribución y variabilidad genética está estrechamente relacionada con regiones geográficas y grupos étnicos [37]. En relación con el presente estudio la variabilidad génica de la G6PD fue baja en los 320 exomas completos de pacientes sin diagnóstico clínico y/o paraclínico pertenecientes al suroccidente colombiano, con un total de 36 variantes, de las cuales 29 variantes no han sido reportadas previamente, 4 variantes presentaron un significado clínico de benigno/probablemente benigno y 3 variantes presentaron un significado clínico incierto.
La variante de significado incierto c.292G>A, p.Val98Met, también conocida como la variante G6PD A- (rs1050828), [38], ha sido descrita en las poblaciones africanas como productora del 10 al 60% de la actividad enzimática normal. Presentó una frecuencia alélica de 0,025 en la población del suroeste colombiano. Sin embargo, la variante homogénea p.Val98Met no es suficiente para causar una deficiencia enzimática y requiere la asociación con la variante c.466A>G, p.Asn156Asp. [39]. Esta variante presentó una frecuencia de 0.043 en el presente estudio, siendo una frecuencia mayor a la reportada para Latinoamérica en bases de datos como gnomAD y 1000 genomas (Tabla 5).
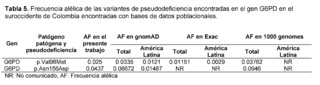
Los estudios de Valencia SH et al. también evaluaron la prevalencia de la deficiencia de G6PD en 426 individuos mediante análisis de actividad enzimática y genotipificación en regiones endémicas de malaria de la costa pacífica colombiana [40]. En estas regiones localizadas en el suroccidente colombiano, Valencia SH et al. reportaron la existencia de las variantes G6PDD A-; p.Val98Met y p.Asn156Asp con deficiencia enzimática de severa a intermedia en 6.62% de los hombres hemicigotos y 4.64% en las mujeres; y junto con la variante A+ que sólo incluye; c.376 A>G, reportó una deficiencia enzimática intermedia en cinco hombres hemicigotos y una mujer heterocigota, siendo la mayor proporción de individuos con haplotipos A- y A+ de G6PD, provenientes de Tumaco y Buenaventura, regiones relacionadas con la población de estudio.
Los estudios de Vela-Amieva Met al., en una cohorte de 81 recién nacidos mexicanos, en un programa de tamizaje neonatal para G6PDd encontraron nueve genotipos G6PDd diferentes; siendo el G6PD A- p.Val98Met y p.Asn156Asp los más frecuentes (60. 5%), demostraron que la misma variante de G6PD podía causar una actividad enzimática residual muy variable, cambiando el curso del espectro fenotípico incluso desde el primer día de vida, ya que el porcentaje de actividad residual media de G6PD en pacientes hospitalizados en el periodo neonatal con o sin ictericia neonatal variaba entre el 29 y el 34% [42].
Los ensayos in-silico realizados por Batista da Rocha J. et al. evaluaron el impacto funcional de las variantes africanas, p.Val98Met y p.Asn156Asp, utilizando técnicas bioinformáticas estructurales. Han descrito cambios significativos en los residuos del dominio de unión catalítica del NADP+, en términos de dirección e impacto, destacando que las diferencias son de orden adictivo, en relación a la unión en NADP+ y G6PD, encontrando cambios mayores con la variante p.Val98Met en contraste con la variante p.Asn156Asp. Sin embargo, la presencia de ambas variantes, el genotipo G6PD A- mostró una mayor inestabilidad en la función enzimática en comparación con la variante p.Val98Met sola [43].
CONSLUSION
En la población estudiada se encontró una alta frecuencia de las variantes p.Val98Met y p.Asn156Asp asociadas con G6PDd, la cual tiene un significado clínico incierto en la literatura y su presencia en portadores se asocia con deficiencia de G6PD. Estos hallazgos sugieren que estas variantes en el gen G6PD podrían considerarse un factor de riesgo para la población del suroccidente colombiano. Así mismo, con una menor frecuencia alélica, la variante patogénica p.Ser135Leu en el gen GALT asociada a galactosemia clásica, circula dentro del suroccidente colombiano, sugiriendo que patologías como la galactosemia pueden atribuir a la carga diagnóstica de EIM por carbohidratos dentro del territorio colombiano.
Este trabajo incrementó la información reportada hasta el momento sobre las variantes asociadas a los EIM por carbohidratos, permitiendo futuras investigaciones, reconociendo el impacto de las variantes patogénicas y pseudodeficiencia que se pueden generar en la sociedad, además contribuyó al conocimiento y creación de bases moleculares sobre los EIM por carbohidratos en el suroccidente colombiano, permitiendo una adecuada consejería genética y educación sobre el riesgo de heredabilidad, realizando medicina anticipatoria y preventiva, acercándonos a la medicina de precisión.
El uso de bases de datos y software bioinformático permitió predecir el significado clínico de las distintas variantes, desde el genotipo hasta el fenotipo. Esta investigación no sólo ofrece una comprensión más profunda de la variabilidad genética en la población estudiada, sino que también pretende ser una referencia valiosa para investigaciones similares en diferentes contextos geográficos. Este enfoque integral puede contribuir al desarrollo de estrategias de salud pública y personalizadas en la identificación y gestión de los EIM de carbohidratos a nivel mundial.
AGRADECIMIENTOS
Gracias a los pacientes participantes, al Instituto de Genética Médica Genómica (GENOMICS), a la Universidad del Valle y al Grupo de Investigación en Enfermedades Metabólicas Congénitas. CONTRIBUCIONES DE AUTOR
JDVB: Investigación, LJMG: Supervisión, Supervisión: JMSS: Supervisión.
Citas
[1] Stenton SL, Kremer LS, Kopajtich R, Ludwig C, Prokisch H. The diagnosis of inborn errors of metabolism by an integrative “multi‐omics” approach: A perspective encompassing genomics, transcriptomics, and proteomics. J Inherit Metab Dis [Internet]. 2020 Jan 25;43(1):25–35. Available from: https://onlinelibrary.wiley.com/doi/10.1002/jimd.12130
[2] Ferreira CR, Rahman S, Keller M, Zschocke J. An international classification of inherited metabolic disorders ( ICIMD ). J Inherit Metab Dis [Internet]. 2021 Jan 27;44(1):164–77. Available from: https://onlinelibrary.wiley.com/doi/10.1002/jimd.12348
[3] Ferguson AM. Chapter 7 - Carbohydrate disorders. In: Garg U, Smith LDBT-B in IE of M, editors. San Diego: Elsevier; 2017. p. 155–66. Available from: http://www.sciencedirect.com/science/article/pii/B9780128028964000134
[4] Ferreira CR, van Karnebeek CDM, Vockley J, Blau N. A proposed nosology of inborn errors of metabolism. Genet Med [Internet]. 2019 Jan 8;21(1):102–6. Available from: http://www.nature.com/articles/s41436-018-0022-8
[5] Guo K, Zhou X, Chen X, Wu Y, Liu C, Kong Q. Expanded Newborn Screening for Inborn Errors of Metabolism and Genetic Characteristics in a Chinese Population. Front Genet [Internet]. 2018 Apr 20;9. Available from: http://journal.frontiersin.org/article/10.3389/fgene.2018.00122/full
[6] Tebani A, Abily-Donval L, Afonso C, Marret S, Bekri S. Clinical Metabolomics: The New Metabolic Window for Inborn Errors of Metabolism Investigations in the Post-Genomic Era. Int J Mol Sci [Internet]. 2016 Jul 20;17(7):1167. Available from: http://www.mdpi.com/1422-0067/17/7/1167
[7] Moreno-Giraldo LJ, Satizábal-Soto JM. Aproximación Diagnóstica de los Errores Innatos del Metabolismo. In: “Estructura del genoma humano y las enfermedades genéticas” Semiología pediatrica. Colombia: Editorial Distribuna; 2021. p. 511–24.
[8] Puentes-Tellez MA, Lerma-Barbosa PA, Garzón-Jaramillo RG, Suarez DA, Espejo-Mojica AJ, Guevara JM, et al. A perspective on research, diagnosis, and management of lysosomal storage disorders in Colombia. Heliyon [Internet]. 2020 Mar;6(3):e03635. Available from: https://linkinghub.elsevier.com/retrieve/pii/S2405844020304801
[9] Congreso de la República de Colombia. Ley 1990 De 2019. 2019;(51):17–2012.
[10] Ministerio de Salud y Protección Social. Resolución 5265 De 2018 Listado de Enfermedades Huérfanas. Minist Salud y Protección Soc [Internet]. 2018;26. Available from: http://achc.org.co/wp-content/uploads/2018/12/RESOLUCIÓN-No.-5265-DE-2018-MINSALUD-LISTADO-ENFERMEDADES-HUÉRFANAS..pdf
[11] Avellaneda LAB. Los Errores Innatos del Metabolismo, fuente inagotable de conocimiento médico por más de un siglo, siguen sin diagnosticarse y tratarse en Colombia. NOVA. 2005;3(3).
[12] Shakiba M, Keramatipour M. Effect of Whole Exome Sequencing in Diagnosis of Inborn Errors of Metabolism and Neurogenetic Disorders. Iran J child Neurol. 2018;12(1):7–15.
[13] González-Lamuño Leguina D, Bóveda Fontán MD, Bueno Delgado M, Gort Mas L, Unceta Suárez M, Morales Conejo M. [The metabolic newborn screening as a healthcare model of the precision medicine. Perspective from the Spanish Association for the Study of Congenital Errors of Metabolism (AECOM).]. Rev Esp Salud Publica [Internet]. 2021 Jan 26;95. Available from: http://www.ncbi.nlm.nih.gov/pubmed/33496277
[14] Ferreira CR, van Karnebeek CDM. Inborn errors of metabolism. In 2019. p. 449–81. Available from: https://linkinghub.elsevier.com/retrieve/pii/B9780444640291000229
[15] Afzal RM, Lund AM, Skovby F. The impact of consanguinity on the frequency of inborn errors of metabolism. Dan Med J [Internet]. 2018 Oct;65(10). Available from: http://www.ncbi.nlm.nih.gov/pubmed/30269750
[16] Guauque-olarte S, Sc B, Patricia Á. DIVERSIDAD Y ESTRUCTURA GENÉTICA DE TRES POBLACIONES AFRODESCENDIENTES DEL SUROCCIDENTE COLOMBIANO A PARTIR DE 8 STR’S. 2010;15:47–60.
[17] Moreno-Giraldo LJ, Satizábal-Soto JM, Sanchéz-Gomez A. Research Article Genomic variability of the mucopolysaccharidosis complex in southwestern Colombia. Genet Mol Res [Internet]. 2020;19(2). Available from: http://www.funpecrp.com.br/gmr/articles/year2020/vol19-2/pdf/gmr18502_-_genomic-variability-mucopolysaccharidosis-complex-southwestern-colombia.pdf
[18] Landrum MJ, Kattman BL. ClinVar at five years: Delivering on the promise. Hum Mutat [Internet]. 2018 Nov 11;39(11):1623–30. Available from: https://onlinelibrary.wiley.com/doi/10.1002/humu.23641
[19] Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R, et al. VarSome: the human genomic variant search engine. Bioinformatics. 2019 Jun;35(11):1978–80.
[20] Li Q, Wang K. InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am J Hum Genet [Internet]. 2017 Feb;100(2):267–80. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0002929717300046
[21] Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med [Internet]. 2015 May 5;17(5):405–23. Available from: http://www.nature.com/articles/gim201530
[22] Andrews C. The Hardy-Weinberg Principle. 2010;10(Nature Education Knowledge):65.
[23] Banford S, McCorvie TJ, Pey AL, Timson DJ. Galactosemia: Towards Pharmacological Chaperones. J Pers Med [Internet]. 2021 Feb 7;11(2):106. Available from: https://www.mdpi.com/2075-4426/11/2/106
[24] Henderson H, Leisegang F, Brown R, Eley B. The clinical and molecular spectrum of galactosemia in patients from the Cape Town region of South Africa. BMC Pediatr [Internet]. 2002 Dec 2;2(1):7. Available from: http://bmcpediatr.biomedcentral.com/articles/10.1186/1471-2431-2-7
[25] Suzuki M, West C, Beutler E. Large-scale molecular screening for galactosemia alleles in a pan-ethnic population. Hum Genet [Internet]. 2001 Aug 1;109(2):210–5. Available from: http://link.springer.com/10.1007/s004390100552
[26] Kumar S U, Kumar D T, R S, Doss C GP, Zayed H. An extensive computational approach to analyze and characterize the functional mutations in the galactose-1-phosphate uridyl transferase (GALT) protein responsible for classical galactosemia. Comput Biol Med [Internet]. 2020 Feb;117:103583. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0010482519304366
[27] Coelho AI, Ramos R, Gaspar A, Costa C, Oliveira A, Diogo L, et al. A frequent splicing mutation and novel missense mutations color the updated mutational spectrum of classic galactosemia in Portugal. J Inherit Metab Dis [Internet]. 2014 Jan 8;37(1):43–52. Available from: http://doi.wiley.com/10.1007/s10545-013-9623-1
[28] Crespo C, Eiroa H, Otegui MI, Bonetto MC, Chertkoff L, Gravina LP. Molecular analysis of GALT gene in Argentinian population: Correlation with enzyme activity and characterization of a novel Duarte-like allele. Mol Genet Metab Reports [Internet]. 2020 Dec;25:100695. Available from: https://linkinghub.elsevier.com/retrieve/pii/S2214426920301415
[29] Chande AT, Nagar SD, Rishishwar L, Mariño-Ramírez L, Medina-Rivas MA, Valderrama-Aguirre AE, et al. The Impact of Ethnicity and Genetic Ancestry on Disease Prevalence and Risk in Colombia. Front Genet [Internet]. 2021 Sep 17;12. Available from: https://www.frontiersin.org/articles/10.3389/fgene.2021.690366/full
[30] Costa-Verdera H, Collaud F, Riling CR, Sellier P, Nordin JML, Preston GM, et al. Hepatic expression of GAA results in enhanced enzyme bioavailability in mice and non-human primates. Nat Commun [Internet]. 2021 Dec 4;12(1):6393. Available from: https://www.nature.com/articles/s41467-021-26744-4
[31] Yang C-C, Chien Y-H, Lee N-C, Chiang S-C, Lin S-P, Kuo Y-T, et al. Rapid progressive course of later-onset Pompe disease in Chinese patients. Mol Genet Metab [Internet]. 2011 Nov;104(3):284–8. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1096719211001971
[32] Reuser AJJ, Ploeg AT, Chien Y, Llerena J, Abbott M, Clemens PR, et al. GAA variants and phenotypes among 1,079 patients with Pompe disease: Data from the Pompe Registry. Hum Mutat [Internet]. 2019 Nov 7;40(11):2146–64. Available from: https://onlinelibrary.wiley.com/doi/10.1002/humu.23878
[33] Burton BK, Charrow J, Hoganson GE, Fleischer J, Grange DK, Braddock SR, et al. Newborn Screening for Pompe Disease in Illinois: Experience with 684,290 Infants. Int J Neonatal Screen [Internet]. 2020 Jan 21;6(1):4. Available from: https://www.mdpi.com/2409-515X/6/1/4
[34] Ficicioglu C, Ahrens-Nicklas RC, Barch J, Cuddapah SR, DiBoscio BS, DiPerna JC, et al. Newborn Screening for Pompe Disease: Pennsylvania Experience. Int J Neonatal Screen [Internet]. 2020 Nov 13;6(4):89. Available from: https://www.mdpi.com/2409-515X/6/4/89
[35] Palmio J, Udd B. Borderlines between Sarcopenia and Mild Late-Onset Muscle Disease. Front Aging Neurosci [Internet]. 2014 Sep 29;6. Available from: http://journal.frontiersin.org/article/10.3389/fnagi.2014.00267/abstract
[36] de Las Heras J, Cano A, Vinuesa A, Montes M, Unceta Suarez M, Arza A, et al. Importance of Timely Treatment Initiation in Infantile-Onset Pompe Disease, a Single-Centre Experience. Child (Basel, Switzerland). 2021 Nov;8(11).
[37] Sathupak S, Leecharoenkiat K, Kampuansai J. Prevalence and molecular characterization of glucose-6-phosphate dehydrogenase deficiency in the Lue ethnic group of northern Thailand. Sci Rep [Internet]. 2021 Dec 3;11(1):2956. Available from: http://www.nature.com/articles/s41598-021-82477-w
[38] Walakira A, Tukwasibwe S, Kiggundu M, Verra F, Kakeeto P, Ruhamyankaka E, et al. Marked variation in prevalence of malaria-protective human genetic polymorphisms across Uganda. Infect Genet Evol [Internet]. 2017 Nov;55:281–7. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1567134817303271
[39] Ouattara AK, Yameogo P, Traore L, Diarra B, Assih M, Compaore TR, et al. Prevalence, genetic variants and clinical implications of G-6-PD deficiency in Burkina Faso: a systematic review. BMC Med Genet [Internet]. 2017 Dec 23;18(1):139. Available from: https://bmcmedgenet.biomedcentral.com/articles/10.1186/s12881-017-0496-2
[40] Valencia SH, Ocampo ID, Arce-Plata MI, Recht J, Arévalo-Herrera M. Glucose-6-phosphate dehydrogenase deficiency prevalence and genetic variants in malaria endemic areas of Colombia. Malar J [Internet]. 2016 Dec 26;15(1):291. Available from: https://malariajournal.biomedcentral.com/articles/10.1186/s12936-016-1343-1
[41] Carmona-fonseca J, Alvarez G, Rios A, Vasquez MF. Deshidrogenasa en hombres sanos y en pacientes maláricos ; Turbo ( Antioquia , Colombia ) dehydrogenase in healthy men and. Rev Bras Epidemiol. 2008;11(2):252–65.
[42] Vela-Amieva M, Alcántara-Ortigoza MA, González-del Angel A, Belmont-Martínez L, López-Candiani C, Ibarra-González I. Genetic spectrum and clinical early natural history of glucose-6-phosphate dehydrogenase deficiency in Mexican children detected through newborn screening. Orphanet J Rare Dis [Internet]. 2021 Dec 26;16(1):103. Available from: https://ojrd.biomedcentral.com/articles/10.1186/s13023-021-01693-9
[43] Batista da Rocha J, Othman H, Hazelhurst S. Molecular dynamics of G6PD variants from sub-Saharan Africa. Biochem Biophys Reports [Internet]. 2022 Jul;30:101236. Available from: https://linkinghub.elsevier.com/retrieve/pii/S240558082200036
