
Caracterización y prevalencia de comorbilidades en pacientes pediátricos con Síndrome de Down de Republica Dominicana
Characterization and prevalence of comorbidities in pediatric patients with Down syndrome in the Dominican RepublicKatlin De La Rosa Poueriet1
 , Andrea Irina Servalle Mella2, Bary Bigay Mercedes3
, Andrea Irina Servalle Mella2, Bary Bigay Mercedes3 1. Hospital Pediátrico Dr Hugo Mendoza, Santo Domíngo, República Dominicana; 2. Universidad Iberoamericana, Santo Domíngo, República Dominicana; 3. Instituto ChromoMED, Santo Domíngo, República Dominicana;
Descargas
Resumen
Este estudio analiza la prevalencia y características de comorbilidades en 534 niños con Síndrome de Down (SD) en la República Dominicana, entre 2018 y 2022. La investigación revela una detección temprana del SD, con una distribución equitativa de género. La mayoría de los casos resultaron de la no disyunción, con una asociación significativa entre la edad materna avanzada y el aumento en el riesgo de SD. Alrededor del 62.2% de los niños presentaron comorbilidades, con condiciones cardíacas como las más prevalentes, seguidas por comorbilidades endocrinas y neurológicas, principalmente hipotiroidismo y trastornos epilépticos. Las afecciones oftálmicas y otorrinolaringológicas también fueron comunes, destacando el estrabismo y la hipoacusia. Los hallazgos enfatizan la necesidad de un manejo temprano e integral adaptado a las características individuales y regionales de los pacientes con SD.
La Trisomía 21, conocida como Síndrome de Down (SD), se caracteriza por una anormalidad cromosómica resultante de la presencia adicional del cromosoma 21, constituyendo la aneuploidía autosómica más frecuentemente observada y compatible con la supervivencia postnatal, además de ser una de las entidades genéticas de mayor complejidad [1]. La incidencia global estimada del Síndrome de Down se sitúa en aproximadamente 1 por cada 1000 nacimientos vivos [2].
Existen tres fenotipos citogenéticos responsables de la trisomía 21: (1) la Trisomía 21 libre por no disyunción, presente en el 95% de los casos y caracterizada por un total de 47 cromosomas; (2) la translocación robertsoniana, con un cromosoma 21 suplementario adherido a otro cromosoma, representando el 4% de los casos; y (3) el mosaicismo de trisomía 21 junto con la trisomía parcial 21, que conforman el 1% de los casos y generalmente se correlacionan con manifestaciones del SD de intensidad variable. El excedente de material genético induce a una serie de manifestaciones clínicas distintivas y incrementa el riesgo de desarrollar diversas comorbilidades [3].
Diversas patologías médicas presentan una mayor prevalencia en individuos con SD comparado con la población general, algunas de las cuales requieren intervenciones inmediatas tras el nacimiento, mientras que otras ameritan una supervisión continua a lo largo de la vida. Se estima que aproximadamente el 75% de los individuos con SD manifiestan al menos una comorbilidad de diverso grado de severidad [4]. A pesar de la variabilidad reportada en la literatura respecto a las comorbilidades más prevalentes en la trisomía 21, se observa una preponderancia de anomalías cardíacas, aproximadamente en el 44% de los casos [5].
Se postula que la diversidad en los tipos de comorbilidades vinculadas a la trisomía 21 y sus manifestaciones específicas difieren significativamente a nivel mundial, sugiriendo una influencia de factores genéticos, sociodemográficos y geográficos [6]. En consecuencia, diversos países han efectuado estudios para determinar con precisión la prevalencia y las variantes de comorbilidades asociadas a la trisomía 21 en sus respectivas poblaciones.
La prevalencia y el manejo de las condiciones de salud en individuos con Síndrome de Down (SD) han sido objeto de numerosos estudios a lo largo de los años. Estos estudios han proporcionado una comprensión más profunda de las necesidades médicas y sociales de esta población. Según Chicoine et al. [7], las personas con SD en los Estados Unidos presentan una alta prevalencia de enfermedades comunes, lo que resalta la importancia de un enfoque centrado en el paciente para la atención médica. Lagan et al. [8] destacaron la implicación multiorgánica y la necesidad de una gestión integral en niños con SD, subrayando la diversidad de necesidades médicas que requieren atención.
Estudios anteriores, como el de Parker et al. [9], han actualizado las estimaciones de prevalencia de nacimientos con SD, destacando un patrón de nacimiento consistente y la necesidad de recursos continuos para estos individuos. En consonancia con esto, Roizen y Patterson [10] proporcionaron una revisión exhaustiva sobre el SD, mientras que Weijerman y de Winter[11] enfatizaron en la importancia de la práctica clínica y el cuidado específico para los niños con SD.
La mortalidad temprana y las causas de muerte en personas con SD también han sido objeto de estudio, como indican O'Leary et al. [12], quienes señalan que las condiciones comórbidas pueden contribuir significativamente a la morbilidad y mortalidad en esta población. Además, Zigman y Lott [13] exploraron la relación entre el SD y la enfermedad de Alzheimer, destacando la importancia de entender los riesgos neurológicos asociados con el SD.
La epidemiología del SD, discutida por Sherman et al. [14], ha proporcionado información valiosa sobre las tasas y tendencias de incidencia. Además, Korenberg et al. [15] investigaron los fenotipos del SD relacionados con el desequilibrio cromosómico, mientras que Gardiner et al. [16] discutieron la utilidad de los modelos de ratón en la investigación del SD. El trabajo de Hassold y Hunt [17] sobre la génesis de la aneuploidía humana ofrece una visión crítica de los errores meióticos que conducen al SD. Finalmente, Capone et al. [18] abordaron la necesidad de un enfoque comprensivo hacia el cuidado de adultos con SD. Estos estudios colectivamente proporcionan una base sólida para comprender los desafíos y avances en el cuidado y manejo de personas con Síndrome de Down.
En la República Dominicana, se han realizado estudios no publicados centrados en malformaciones cardíacas congénitas en la población pediátrica afectada por SD, pero aún se carece de un registro exhaustivo y accesible al público sobre las comorbilidades presentes en este grupo demográfico. La presente investigación tiene como finalidad recolectar y actualizar datos sobre la prevalencia de comorbilidades vinculadas a la trisomía 21 en la región dominicana, con el propósito de detallar cada patología y categorizarlas según su severidad o la necesidad de intervención quirúrgica.
Se implementó una investigación observacional, descriptiva y transversal de naturaleza retrospectiva, por medio del análisis de historiales clínicos de sujetos diagnosticados con Síndrome de Down, que recurrieron al departamento de genética médica del Hospital Pediátrico Dr. Hugo Mendoza, República Dominicana, abarcando el intervalo de agosto de 2018 a agosto de 2022.
El proceso de análisis involucró una revisión minuciosa y sistemática de la documentación clínica existente, sin alteraciones ni intervenciones directas de los investigadores sobre los participantes. Posteriormente, los datos obtenidos se consolidaron en una base de datos diseñada específicamente para esta investigación, empleando los registros clínicos digitales de la Clínica de Atención Integral para Pacientes con Síndrome de Down y el Servicio de Genética Médica del mencionado hospital.La metodología para la recolección de datos se centró en la organización manual de la información clave extraída para cada paciente, utilizando Microsoft Office Excel®. Se configuró una base de datos estructurada en cinco segmentos principales: 1) identificación de la historia clínica, 2) información general y sociodemográfica, 3) confirmación diagnóstica de la trisomía 21, 4) identificación de comorbilidades asociadas, y 5) intervenciones quirúrgicas realizadas, subdividiendo cada categoría para una catalogación detallada y precisa.Dado el enfoque observacional y retrospectivo del estudio, no se solicitó la participación de los pacientes, destacando la importancia de la confidencialidad y la protección de datos personales, en consonancia con los lineamientos éticos.
Este proceder asegura la protección de la privacidad y la integridad de los menores, en estricto acatamiento a las directrices del Comité de Ética de Investigación (CEI) del Hospital Dr. Hugo Mendoza, con fines exclusivamente académicos y científicos.La muestra inicial comprendió 2,048 pacientes, de los cuales, tras aplicar los criterios de inclusión y exclusión, se seleccionó una cohorte final de 534 sujetos pediátricos con diagnóstico confirmado de Síndrome de Down. Utilizando técnicas de muestreo probabilístico y estableciendo un margen de error del 1% y un nivel de confianza del 99% mediante el software Raosoft®, se validó la representatividad de la muestra.
Los criterios de inclusión definidos fueron: pacientes pediátricos (menores de 18 años) evaluados en la consulta de Genética Médica del HPHM en el periodo establecido, con un diagnóstico confirmado de Síndrome de Down mediante análisis genético. Se excluyeron aquellos registros con información incompleta, pacientes atendidos fuera del rango temporal definido y diagnósticos basados únicamente en criterios clínicos.
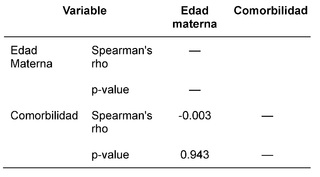
Tras la recopilación y categorización de los datos, se procedió al análisis conforme a los objetivos propuestos, aplicando técnicas estadísticas de distribución de frecuencias, así como medidas de tendencia central y dispersión. Complementariamente, se empleó el software estadístico JASP® para realizar correlaciones mediante el coeficiente de Spearman.
DISCUSION
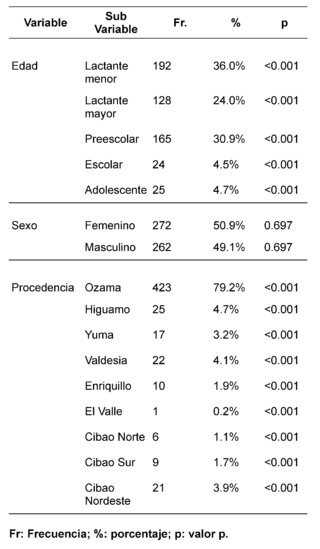
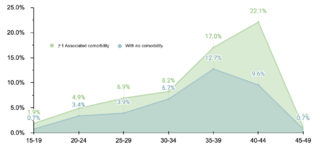
En la investigación se incluyeron 534 sujetos pediátricos diagnosticados con Síndrome de Down (SD) entre agosto de 2018 y agosto de 2022. La distribución por género fue casi equitativa, con un 50.9% (272/534) de participantes femeninos y un 49.1% (262/534) masculinos, evidenciando la ausencia de sesgo de género en la incidencia del SD. La media de edad se estableció en 2.4 años ± 3.1 DE, identificándose una mayor incidencia en el grupo de lactantes menores (1-11 meses), lo que representa un 36% (192/534) de la muestra. Este dato sugiere una temprana detección y abordaje de comorbilidades en el SD, contribuyendo potencialmente a una disminución en la morbimortalidad de esta población.
La evaluación etiológica reveló que la mayor parte de los casos de SD (99%, 529/534) se debieron a no disyunción, seguidos por translocación Robertsoniana (0.6%, 3/534) y mosaicismo (0.4%, 2/534). Estos hallazgos están en consonancia con distribuciones etiológicas preestablecidas. La edad materna promedio en el momento del parto fue de 34.8 años ± 6.7 DE, con una frecuencia mayor en el rango de 40-44 años. Este patrón reafirma la asociación entre mayor edad materna y un incrementado riesgo de diagnóstico de SD.
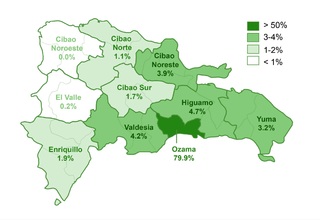
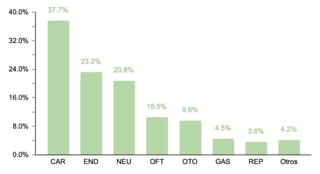
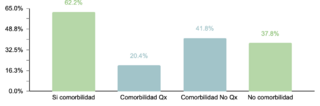
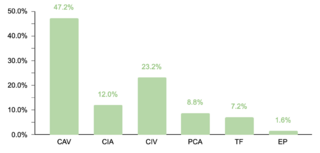
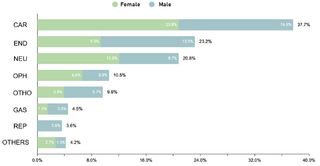
El estudio identificó que un 62.2% (332/534) de la muestra manifestó una o más comorbilidades asociadas al SD, una proporción menor en comparación con estudios realizados en otras regiones. Se observó que un 20.4% (109/332) de los pacientes con comorbilidades requerían intervenciones quirúrgicas. La prevalencia de comorbilidades cardíacas fue del 37.7% (125/332), con la comunicación atrioventricular (CAV) siendo la más frecuente. Aunque este porcentaje es comparable con datos de la India, es menor en relación con cifras reportadas en EE.UU., España, Chile y Bangladesh.
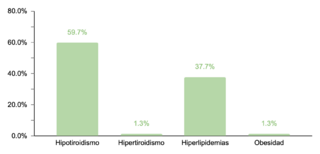
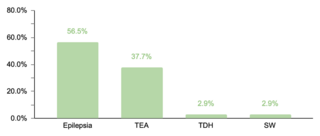
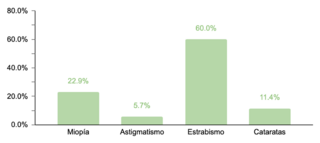
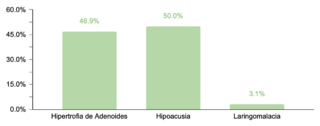
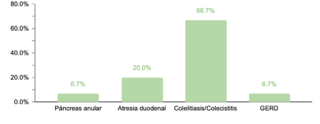
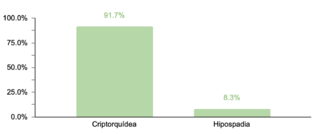
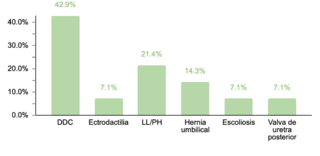
En cuanto a comorbilidades endocrinas, un 23.2% (77/332) de los pacientes presentaron alteraciones, siendo el hipotiroidismo la más común. Respecto a las comorbilidades neurológicas, un 20.8% (69/332) de los casos fue diagnosticado, destacando los trastornos epilépticos como la condición predominante. Se encontró que un 10.5% (35/332) de los niños tenía afecciones oftálmicas, principalmente estrabismo, atribuible a la hipotonía muscular generalizada típica en SD. Las comorbilidades otorrinolaringológicas afectaron al 9.6% (32/332) de la muestra, con la hipoacusia como la condición más común. Un menor porcentaje mostró comorbilidades gastrointestinales y del sistema reproductor, siendo la colelitiasis y la criptorquidia las más prevalentes, respectivamente.
La investigación resalta la variabilidad de las comorbilidades asociadas al SD y subraya la importancia de un abordaje integral y precoz en el manejo de estos pacientes, adaptado a las necesidades específicas de cada caso y basado en un entendimiento profundo de la prevalencia y naturaleza de las comorbilidades en diferentes contextos geográficos y demográficos.
Este estudio demostró que una proporción considerable de la población pediátrica con Síndrome de Down (SD) presenta múltiples comorbilidades, con un énfasis particular en las alteraciones cardíacas, destacando la Comunicación Atrioventricular (CAV) como la más común. A pesar de que el género no influyó en la incidencia del SD, se identificó una tendencia específica hacia determinadas comorbilidades reproductivas, oftálmicas y gastrointestinales en función del género de los pacientes. Además, se reafirmó que la edad materna avanzada es un factor de riesgo significativo para el desarrollo de SD, ya que una amplia mayoría de los casos estudiados correspondieron a madres mayores de 35 años.
La necesidad de intervenciones quirúrgicas en una parte significativa de los pacientes con comorbilidades subraya la complejidad de las condiciones asociadas y la importancia de un manejo médico especializado. Los hallazgos de este estudio refuerzan la necesidad de una estrategia de atención médica personalizada para la población pediátrica con SD en la República Dominicana, diferenciada de los enfoques aplicados a la población general, con el fin de mejorar la detección temprana y el tratamiento de comorbilidades, reduciendo así la morbimortalidad asociada. Este enfoque debe adaptarse a las necesidades específicas de cada paciente, teniendo en cuenta las particularidades locales y demográficas para garantizar una atención óptima y oportuna.
Citas
[1] Antonarakis SE, Skotko BG, Rafii MS, Strydom A, Pape SE, Bianchi DW, et al. Down syndrome. Nat Rev Dis Primers. 2020 Feb 6;6(1):9. doi: 10.1038/s41572-019-0143-7.
[2] United Nations. World down syndrome day [Internet]. United Nations; 2023 [cited 2023 Feb 9]. Available from: https://www.un.org/en/observances/down-syndrome-day.
[3] Bull MJ. Down Syndrome. In: Ropper AH, editor. N Engl J Med. 2020 Jun 11;382(24):2344–52. doi: 10.1056/NEJMra1706537.
[4] Heinke D, Isenburg JL, Stallings EB, Short TD, Le M, Fisher S, et al. Prevalence of structural birth defects among infants with Down syndrome, 2013–2017: A US population‐based study. Birth Defects Res. 2021 Jan 15;113(2):189–202. doi: 10.1002/bdr2.1854.
[5] Stoll C, Dott B, Alembik Y, Roth MP. Associated congenital anomalies among cases with Down syndrome. Eur J Med Genet. 2015;58(12):674–80. doi: 10.1016/j.ejmg.2015.11.003.
[6] De Rubens Figueroa J, del Pozzo Magaña B, Pablos Hach JL, Calderón Jiménez C, Castrejón Urbina R. Malformaciones cardíacas en los niños con síndrome de Down. Rev Esp Cardiol. 2003;56(9):894–9. doi: 10.1157/13051617.
[7] Chicoine B, Rivelli A, Fitzpatrick V, Chicoine L, Jia G, Rzhetsky A. Prevalence of Common Disease Conditions in a Large Cohort of Individuals With Down Syndrome in the United States. J Patient-Cent Res Rev. 2021 Apr 19;8(2):86–97. doi: 10.17294/2330-0698.1824.
[8] Lagan N, Huggard D, Mc Grane F, Leahy TR, Franklin O, Roche E, et al. Multiorgan involvement and management in children with Down syndrome. Acta Paediatr. Blackwell Publishing Ltd; 2020; 109:1096–111. doi: 10.1111/apa.15153.
[9] Parker SE, Mai CT, Canfield MA, Rickard R, Wang Y, Meyer RE, et al. Updated National Birth Prevalence estimates for selected birth defects in the United States, 2004-2006. Birth Defects Res A Clin Mol Teratol. 2010 Dec;88(12):1008-16. doi: 10.1002/bdra.20735.
[10] Roizen NJ, Patterson D. Down's syndrome. Lancet. 2003 Apr 12;361(9365):1281-9. doi: 10.1016/S0140-6736(03)12987-X.
[11] Weijerman ME, de Winter JP. Clinical practice. The care of children with Down syndrome. Eur J Pediatr. 2010 Dec;169(12):1445-52. doi: 10.1007/s00431-010-1253-0.
[12] O'Leary L, Hughes-McCormack L, Dunn K, Cooper SA. Early death and causes of death of people with Down syndrome: A systematic review. J Appl Res Intellect Disabil. 2018 Jul;31(4):687-708. doi: 10.1111/jar.12446.
[13] Zigman WB, Lott IT. Alzheimer's disease in Down syndrome: Neurobiology and risk. Ment Retard Dev Disabil Res Rev. 2007;13(3):237-46. doi: 10.1002/mrdd.20163
[14] Sherman SL, Allen EG, Bean LH, Freeman SB. Epidemiology of Down syndrome. Ment Retard Dev Disabil Res Rev. 2007;13(3):221-7. doi: 10.1002/mrdd.20157.
[15] Korenberg JR, Chen XN, Schipper R, Sun Z, Gonsky R, Gerwehr S, et al. Down syndrome phenotypes: The consequences of chromosomal imbalance. Proc Natl Acad Sci U S A. 1994 May 24;91(11):4997-5001. doi: 10.1073/pnas.91.11.4997.
[16] Gardiner KJ, Fortna A, Bechtel L, Davisson MT. Mouse models of Down syndrome: How useful can they be? Comparison of the gene content of human chromosome 21 with orthologous mouse genomic regions. Gene. 2003 Mar 6; 318:137-47. doi: 10.1016/S0378-1119(03)00769-8.
[17] Hassold T, Hunt P. To err (meiotically) is human: The genesis of human aneuploidy. Nat Rev Genet. 2001 Apr;2(4):280-91. doi: 10.1038/35066065.
[18] Capone GT, Chicoine BA, Bulova P. Adults with Down Syndrome: A comprehensive approach to care. Curr Opin Psychiatry. 2018 Mar;31(2):113-120. doi: 10.1111/jir.12588
