
Cromoanagénesis y Eritrodermia como claves diagnósticas en un paciente en estado crítico
Chromoanagenesis and Erythroderma as diagnostic clues in a critically ill patientEnrique Daniel Austin-Ward 1,2,3
 , Nicolás Hurtado4, Nelson Santamaría5, Alexis Pinto6, German Espino7,8,9
, Nicolás Hurtado4, Nelson Santamaría5, Alexis Pinto6, German Espino7,8,91. Instituto de Investigaciones Científicas Avanzadas y Servicios de Alta Tecnología - INDICASAT, Panamá, Rep de Panamá; 2. Departamento de Genética Clínica, Hospital Pacífica Salud, Panamá, Rep de Panamá; 3. Instituto de Genética Médica y Genómica, Caja de Seguro Social Panamá; 4. Unidad de Nefrología y Terapia Intensiva, Hospital Santa Fe, Panamá, Rep de Panamá; 5. Unidad de Neumología, Hospital Santa Fe, Panamá, Rep de Panamá; 6. Unidad de Hematología, Hospital Santa Fe, Panamá, Rep de Panamá; 7. Departamento de Hematología, Hospital Santo Tomás, Panamá, Rep de Panamá; 8. Centro Hemato-Oncológico, Panamá, Rep de Panama; 9. Escuela de Medicina, Universidad de Panamá, Panamá, Rep de Panamá;
Descargas
Resumen
Reportamos el caso de una paciente con un cuadro grave de falla multisistémica en la cual se detectaron cambios cromosómicos masivos en estado de mosaico, que se consideran estarían asociados a la posible patología de fondo: un síndrome linfoproliferativo de Células T. La paciente también presentaba un cuadro de Eritrodermia de causa desconocida instaurado varios años antes, con un declive progresivo de su condición física. El espectro de las manifestaciones clínicas se presenta entonces como un cuadro cutáneo inespecífico paraneoplásico de varios años de evolución y hallazgos de cromoanagénesis en la última fase de un proceso mórbido linfoproliferativo. Las nuevas técnicas de análisis genético permiten obtener datos para aclarar los procesos diagnósticos e identificar una desorganización grave de la información genética que se considera responsable de cambios neoplásicos.
Reporte del Caso
Se trata de una paciente femenina de 69 años, tercera hija de padres no consanguíneos, e historia familiar de una hermana con diagnóstico de Lupus Eritematoso Sistémico (LES), quien se encontraba en la Unidad de Cuidados Intensivos, presentando enfermedad sistémica y complicaciones que requirieron hospitalizaciones recurrentes durante tres meses, con empeoramiento progresivo. La paciente presentaba evidencias de hiperinflamación con aumento de la Velocidad de Eritro-Sedimentación (VES) y de la proteína C reactiva.
La tomografía de tórax revelaba áreas de fibrosis pulmonar e imágenes en vidrio esmerilado. Presentaba además bicitopenia con anemia severa y trombocitopenia moderada, infecciones recurrentes de difícil manejo, anasarca, derrame pleural y falla orgánica progresiva. La presencia de múltiples adenopatías hacía sospechar un Síndrome Linfoproliferativo, pero las biopsias de ganglio y médula ósea practicadas hasta ese momento habían resultado negativas por Linfoma, y sólo se presentaban con un hallazgo de displasia moderada de la serie blanca, sin otras conclusiones definitivas.
En este estado crítico, y sin un diagnóstico etiológico, se solicitó evaluación por Genética quien recomienda realizar una prueba de Genoma Completo buscando datos relacionados con alteración oncogenética en la línea germinal, en la inmunidad innata u otra condición genética predisponente, dado que no se disponía de especímenes para otro tipo de estudio molecular. La paciente fallece a los pocos días, con resultados de la prueba genética pendientes.
Antes de la situación clínica actual, la paciente tenía antecedentes de una afección que comenzó aproximadamente hacía siete años, siendo evaluada por Hematolgía por la presencia de un ganglio retroauricular derecho no doloroso, sin síntomas B (fiebre, sudoración, pérdida de peso), ni otros hallazgos en los exámenes de laboratorio y de imagen, ni evidencia de visceromegalias ni de otras adenopatías. Además, la paciente presentaba en ese momento eritema en superficies extensas del cuerpo, que incluían cara, tronco y extremidades (Figura 1), asociado a prurito generalizado acompañado de sensación de ardor y alodinia, áreas de descamación en los pies y en otras partes del cuerpo y edema de extremidades inferiores. Un diagnóstico de Eritrodemia fue realizado, y aunque se descartaron las causas más frecuentes, la paciente abandonó la evaluación por Dermatología sin una conclusión y siguió tratamientos de medicina alternativa con mejoría parcial del cuadro cutáneo.
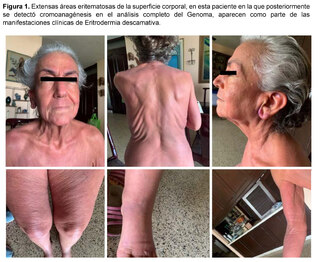
Seis años más tarde la paciente regresa a la Consulta de Hematología con cuadro de adenopatías en cuello, axila, hepatoesplenomegalia, historia de candidiasis recurrente y la persistencia de la Eritrodermia, instalándose el cuadro de edema generalizado, efusión pleural y falla orgánica progresiva ya descrito, con un período prolongado de hospitalización y el desenlace comentado.
Pruebas Moleculares
A partir de muestra de sangre periférica seca en papel filtro, se realizó CentoGenome® MOx 1.0 Solo: El ADN genómico fue fragmentado enzimáticamente y marcado con secuencias adaptadoras compatibles con Ilumina. Las librerías fueron secuenciadas desde ambos extremos (paired-end) en una plataforma Illumina para generar una profundidad de cobertura media de ~30x. Se aplicó un proceso bioinformático basado en el proceso DRAGEN de Illumina, así como el proceso bioinformático interno de CENTOGENE. Las lecturas fueron alineadas con el ensamblaje del genoma Genome Reference Consortium Human Build 37 (GRCh37/hg19), así como a la secuencia de referencia de Cambridge (revised Cambridge Referece Sequence; rCRS) del ADN mitocondrial humano (NC_012920). Las variantes de secuencia (SNVs/indels) y las variantes en el número de copia (CNVs) fueron llamadas usando el algoritmo DRAGEN, Manta y propio. Se evaluaron todas las variantes con una frecuencia alélica minoritaria (MAF) inferior al 1% en la base de datos gnomAD, y las variantes causantes de enfermedad reportadas en HGMD®, en ClinVar o en el Biodatabank de CENTOGENE. Aunque la evaluación se centró en los exones codificantes y las regiones intrónicas flanqueantes, el gen completo se interrogó en busca de variantes candidatas con una asociación plausible con el fenotipo. Se consideraron todos los patrones potenciales de modo de herencia. Además, se utilizaron los antecedentes familiares y la información clínica proporcionada para evaluar las variantes identificadas con respecto a su patogenicidad y causalidad de la enfermedad. Las variantes se clasifican en cinco clases (patogénica, probablemente patogénica, VUS, probablemente benigna y benigna) siguiendo las directrices del ACMG para la clasificación de variantes además de las recomendaciones de ClinGen. Se informaron todas las variantes relevantes relacionadas con el fenotipo del paciente. Las variantes benignas y probablemente benignas no son reportadas. Para CentoGenome® MOx, si el análisis bioquímico es aplicable, este se realiza tras la detección de variantes relevantes mediante secuenciación en genes específicos. Esto mejora el diagnóstico de trastornos metabólicos, optimiza la clasificación de variantes y ayuda a determinar la contribución final al fenotipo; la lista de ensayos de actividad enzimática y biomarcadores se puede obtener en www.centogene.com/mox. Las CNVs de significado incierto sin aparente relación con el fenotipo del paciente no se informaron. Las variantes mitocondriales con niveles de heteroplasmia del 15% o mayores son reportadas. Para la detección de SNV e indels en las regiones seleccionadas para el análisis posterior, se logra una sensibilidad del 99.9%, una especificidad del 99.9% y una precisión del 99.9%. El software de detección de CNVs tiene una sensibilidad de más del 95%. CENTOGENE ha establecido estrictos criterios de calidad y procesos de validación para las variantes detectadas por NGS. Las variantes con baja calidad de secuenciación y/o cigosidad poco clara fueron confirmadas mediante métodos ortogonales. En consecuencia, se garantizó una especificidad > 99.9% para todas las variantes reportadas. El cribado de expansiones de repeticiones se realizó con el algoritmo ExpansionHunter para los siguientes genes: AR, ATN1, ATXN1, ATXN2, ATXN3, ATXN7, ATXN8OS, ATXN10, CACNA1A, CNBP, CSTB, C9ORF72, DMPK, FMR1, FXN, HTT, JPH3, NOP56, PABPN1, PHOX2B, PPP2R2B, PRNP y TBP. Los resultados técnicos del cribado de expansión de repeticiones se correlacionaron con la información clínica proporcionada. Cualquier expansión repetida llamada y considerada relevante para el fenotipo sería confirmada por un método ortogonal. El cribado de GBA1 se realizó mediante el algoritmo Gauchian para detectar eventos de recombinación que afectan a la región que abarca los exones 9-11 (NM_000157.3), una región que tiene la mayor homología con GBAP1. Cualquier evento de recombinación detectado se informaría solo cuando se considere relevante para el fenotipo. El cribado de atrofia muscular espinal (AME) se realizó utilizando el algoritmo SMN Caller para detectar el número de copias del gen SMN1. Cualquier CNV detectada solo se confirmaría mediante un método ortogonal y se informaría cuando se considere relevante para el fenotipo. El cribado de disomía uniparental (UPD) se realizó utilizando un algoritmo interno para errores de herencia mendeliana (MIE) para detectar regiones de homocigosidad (ROH) para las conocidas regiones cromosómicas clínicamente relevantes (6q24, 7, 11p15.5, 14q32, 15q11q13, 20q13 y 20).
CentoArray®: Para este análisis se utilizó el kit Infinium™ Global Diversity Array with Cytogenetics (Illumina). Este kit contiene ~1,8 millones de marcadores SNP distribuidos a lo largo de todo el genoma, en particular para más de 4,800 genes asociados a enfermedad con una cobertura exónica del 99.9%, lo que permite la detección de variaciones en el número de copias (CNV). El ADN genómico se amplificó, fragmentó e hibridó en el array de acuerdo con las instrucciones del fabricante. Los resultados se analizaron utilizando el software NxClinical (BioDiscovery). Se reportaron CNVs que exceden un tamaño de 50 kb para pérdidas y 200 kb para ganancias. Las pérdidas identificadas por debajo de los umbrales dados solo se informaron si se observó una correlación fenotípica clara con los genes afectados. Los resultados se interpretaron utilizando las bases de datos DGV, DECIPHER, ClinGen y recursos adicionales disponibles. Las CNV se clasifican en cinco clases (patogénicas, probablemente patogénicas, VUS, probablemente benignas y benignas) según las recomendaciones del ACMG para la clasificación de variantes. Las CNV benignas y probablemente benignas no fueron reportadas. Las CNV se evaluaron en función del motivo de consulta del paciente para esta prueba y la información clínica proporcionada. El informe completo de las CNV heterocigotas en el contexto de enfermedades recesivas está fuera del alcance del uso previsto para esta prueba. Por lo tanto, es posible que no se revele el estado del portador. Cualquier preocupación clínica por trastornos recesivos debe comunicarse al laboratorio de notificación para su adecuada consideración. De acuerdo con los requisitos reglamentarios en los análisis prenatales, CENTOGENE solo informa CNV patogénicas y probablemente patogénicas de alta penetrancia. Este array permite el análisis de regiones con ausencia de heterocigosidad (AOH). La presencia de AOH en múltiples cromosomas puede ser compatible con la herencia de un ancestro común.
Resultados
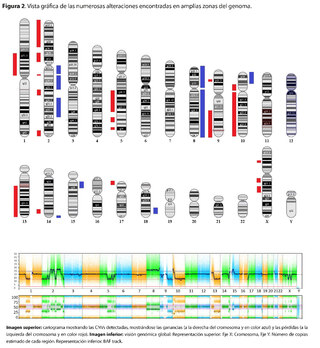
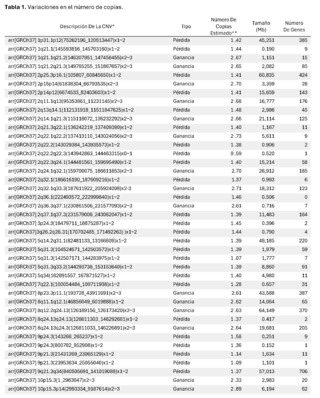
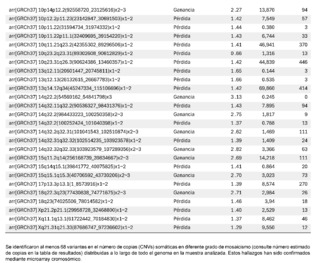
Se identificó la presencia de grandes variantes en el número de copia (CNVs), distribuidos a lo largo de todo el genoma en diferentes grados de mosaicismo en la muestra analizada y, por lo tanto, consideradas de origen somático y no germinal (Ver tabla 1) y confirmados mediante microarray cromosómico (Ver figura 2), y que son compatibles con un proceso de cromoanagénesis, sugiriéndose por parte del laboratorio genético realizar una evaluación hemato oncológica, que en nuestro contexto de sospecha clínica, se trataría de un trastorno linfoproliferativo de fondo.
Discusión
La evaluación rutinaria del cariotipo como fuente de datos inicial para resolver cuadros clínicos, nos transmite en la imagen frecuente de los cromosomas, la impresión de estabilidad de la información genética, en la que las alteraciones en número y estructura se detectan como hallazgos esporádicos. Sólo en las evaluaciones cariotípicas de médula ósea uno está más preparado para observar cambios estructurales y numéricos graves concurriendo en un mismo paciente.
Aunque sabemos que esta imagen de estabilidad muchas veces no corresponde a la realidad última y que las técnicas de CGH (Comparative Genome Hybridization – Hibridización Comparativa del Genoma) nos permiten detectar la presencia de pérdidas o ganancias de material genético no detectables a nivel del Cariotipo y realizar así el diagnóstico de síndromes por microdeleción o mIcroduplicación, los hallazgos en el presente caso con la detección de una debacle cromosómica a gran escala con numerosas variantes en el número de copia (CNVs-Copy Number Variants – Variantes en el Número de Copias), producto de procesos de reorganización cromosómica, es llamativa e inusual.
El término cromoanagénesis (renacimiento cromosómico) fue sugerido por Holland y Cleveland en el año 2012, para describir en su conjunto, una serie de fenómenos consistentes en procesos de reorganización cromosómica masiva, observados gracias a las nuevas tecnologías, como las de secuenciación genómica y el “microarray” utilizados en esta ocasión [1]. Observados y descritos inicialmente por Stephens y cols. [2], estos verdaderos cataclismos en la información genética se han asociado a fenómenos neoplásicos de distinto tipo y a síndromes genético-dismórficos, aunque se han detectado también en la línea germinal de individuos normales, haciéndolos proclives a mayor incidencia de abortos y a descendencia con síndromes genéticos [3].
Recordemos que los genes controlan todo lo relacionado con el diseño y funcionamiento del organismo, incluyendo el control del crecimiento celular y que cuando estos genes se alteran pueden dar lugar al cáncer. Estos genes participan por medio de sus productos, que son las proteínas, en el sistema de comunicación inter e intracelular y que una proteína alterada cuantitativa o cualitativamente constituye el mecanismo inicial para estos trastornos del control del crecimiento celular, ya que el cáncer es una enfermedad genética. El daño genético es la llamada “causa necesaria” y este daño se puede producir por diversos mecanismos, llamados “causas suficientes” [4].
Aparte de las mutaciones puntuales, los rearreglos cromosómicos pueden crear genes de fusión que produzcan “superproteínas”: proteínas con ganancia de función que promuevan un crecimiento celular sin control, o dañar genes esenciales que tengan que ver la reparación del ADN o genes supresores de tumor, que vigilan la aparición de daño genético y tratan de solucionarlo, por lo que se trata en estos dos últimos casos de eventos de pérdida de la función [5].
La cromoanagénesis, la cual incluye tres mecanismos distintos conocidos como cromotripsis, cromoplexia y cromoanasíntesis, nos habla de procesos de remodelación abrupta y cataclísmica del genoma que son propiciados por diferentes circunstancias moleculares, entre la que están involucradas las relacionadas a genes relacionados con el cáncer. La cromotripsis, definida por Pellestor como “un evento mutacional producido por múltiples rupturas de doble hebra, de manera recurrente en un único evento catastrófico entre un número limitado de segmentos cromosómicos y seguido por el reensamblaje de fragmentos de ADN en un orden y orientación al azar para formar cromosomas derivativos” [6], ha sido descrita como el subtipo de rearreglo cromosómico masivo presente de manera importante en los casos de cáncer con cromoanagénesis, aunque, como hemos mencionado, se puede encontrar en otros trastornos genéticos e inclusive en la línea germinal de personas sanas, representando un incremento en el riesgo de abortos o de descendencia con trastornos genéticos [7].”
Por otro lado, la cromoanasíntesis (reconstitución cromosómica), es otro de los mecanismos de cromoanagénesis cuyos efectos son similares a los de la cromotripsis, pero que se ha identificado de forma predominante en cuadros genéticos sindrómicos con dismorfias y discapacidad intelectual [6,8,9], mientras que la cromoplexia (reestructuración cromosómica), pudiese ser el mecanismo predominante en ciertos tipos de cáncer en los que se presente cromoanagénesis como en el de próstata u otros y a diferencia de lo que ocurre en la cromotripsis, los rearreglos presentan pocas o ninguna alteración en el número de copias [6].
Todos estos fenómenos parecen apoyar la teoría de la evolución del cáncer basada en uno o pocos eventos cataclísmicos o teoría del equilibrio puntuado [10], presente también, según las hipótesis, como uno de los mecanismos de especiación en la Naturaleza, en lugar del modelo tradicionalmente invocado consistente en mutaciones acumulativas a largo plazo o gradualista, como nos señala Pellestor, aunque en los casos de cáncer también se ha postulado un modelo mixto [2,6].
La información genética no debe estar ni en exceso ni en déficit, a fin de garantizar la homeostasis básica del sistema de información celular. El desbalance en un solo gen ya es capaz de provocar serios problemas. En este caso, el primero de este tipo reportado en nuestro país, es muy llamativa la notable acumulación de alteraciones cromosómicas complejas, con 68 según el informe obtenido. Para darnos cuenta de la gravedad de la desorganización, se evidenció que está involucró a 14 de los 23 cromosomas (61%). El 66% de estos cambios fueron pérdidas cromosómicas y el 34% ganancias. Los cromosomas más afectados fueron el 2 (26%), el 10 (13%) y el 14 (10%). El rango de las pérdidas de genes oscila entre 0 y 706 y el de las ganancias entre 0 y 387 (Ver Tabla 1).
Aunque no tenemos forma de confirmarlo por medio de estudios adicionales, basándonos en la descripción de las características de los tres mecanismos involucrados en la cromoanagénesis y en qué casos se presentan, los cambios presentados en la Tabla 1 corresponderían a un fenómeno de cromotripsis. En la mayoría de los casos, la desorganización grave del genoma que se presenta en los casos de cromoanagénesis, específicamente del tipo cromotripsis, estarían involucrados hasta entre un 2 y 4% de todos los cánceres y tienen un pronóstico ominoso [6], lo cual pudiese ser usado en el futuro como un marcador en cuanto a la posible evolución de los pacientes. No obstante, no hay datos claros sobre el nivel de tolerancia del sistema y sobre la capacidad de resiliencia para la recuperación, y si existe un umbral de grado de desorganización que, sobrepasado, marcaría un punto de no retorno, aunque se haya instaurado incluso el tratamiento etiológico preciso.
Aunque sin duda en los numerosos casos de cáncer reportados con cromoanagénesis, es probable haya existido la presencia concomitante de cuadros paraneoplásicos, no hemos encontrado reportes de pacientes en que se describa la Eritrodermia como un proceso asociado a cromoanagénesis en el mismo paciente.
La Eritrodermia, como nos comenta Harper-Kirsey, es “una dermatitis grave y potencialmente mortal descrita como un eritema intenso y generalizado que tiende a afectar a más del 90% del área de superficie corporal, con un grado variable de descamación exfoliativa de la piel y puede ser una manifestación de una amplia gama de enfermedades cutáneas y sistémicas, incluidas infecciones, tumores malignos y reacciones de hipersensibilidad a los medicamentos” [11,12,13]. En ese contexto de posibilidades, la Eritrodermia está descrita como un síndrome paraneoplásico de tipo dermatológico presente con frecuencia en neoplasias linfoproliferativas [14].
Los síndromes paraneoplásicos son manifestaciones indirectas del cáncer no atribuibles a la invasión directa del tumor o a efectos de compresión que pueda producir y muchos se expresan antes del diagnóstico del cáncer, por lo que pueden servir de signo centinela. Pueden ser de tipo endocrinológico (secreción inapropiada de la hormona antidiurética, hipercalcemia, Síndrome de Cushing, hipoglicemia), neurológico (encefalitis límbica, degeneración cerebelosa, miastenia gravis), dermatológicos y reumatológicos (dermatomiositis, acantosis nigricans, eritrodermia, osteoartropatía hipertrófica) [14].
En este caso se manejaban sospechas contundentes con respecto a un síndrome linfoproliferativo de células T como diagnóstico de fondo, y nuestra paciente tenía un antecedente de aproximadamente 7 años de evolución con el diagnóstico Eritrodermia descamativa, habiéndose excluido alergias a medicamentos u otras causas frecuentes. Por otro lado, los trastornos linfoproliferativos en algunos casos pueden tener tiempos de incubación superiores a los 5 años [15].
Aunque un diagnóstico definitivo no fue posible en esta ocasión, la presencia de estos cambios genéticos y el padecimiento concomitante de Eritrodermia en esta paciente, refuerzan la hipótesis diagnóstica de un cuadro neoplásico linfoproliferativo. La cromoanagénesis puede ser la causa o la consecuencia manifiesta de un proceso de desorganización masivo en curso, y nos puede hablar sobre el carácter ominoso del padecimiento y sobre su pronóstico, proveyendo a los médicos tratantes con información importante sobre el estatus del paciente en casos extremadamente complejos y de difícil manejo. Cuando los detalles específicos de los algoritmos diagnósticos y de tratamiento de la cromoanagénesis estén disponibles, proveerán de herramientas adicionales para el abordaje oportuno de estas situaciones clínicas.
Nuevas técnicas como el OGM (Optical Genome Mapping – Mapeo Genómico Óptico), apoyarán el hallazgo de estos cambios para una mejor aproximación a estos fenómenos [16].
La valoración del grado de cromoanagénesis tal vez pueda ser utilizado en el futuro como un indicador del grado de desorganización progresiva del sistema o en casos de pacientes con cáncer y que sería necesario revertir para recuperar la homeostasis, como cuando se miden otros parámetros en grados de desorganización multisistémica que nos permiten realizar predicciones en casos específicos [17].
Conclusiones
El uso de las técnicas de análisis completo del Genoma son herramientas de utilidad para aclarar el diagnóstico de pacientes en estado crítico en las que faltan otros datos que les permitan a los médicos tratantes identificar la situación clínica de fondo subyacente a procesos graves. Estas técnicas, junto con las evaluaciones oncogenéticas pueden suministrar piezas claves en el algoritmo diagnóstico y el manejo de estos pacientes.
Es importante además estar muy pendientes de los datos de la historia del paciente, en relación de posibles manifestaciones centinela de tipo dermatológico, reumatológico, neurológico y endocrinológico que no se hayan aclarado completamente.
Citas
[1] Holland AJ, Cleveland DW. Chromoanagenesis and cancer: mechanisms and consequences of localized, complex chromosomal rearrangements. Nat Med. 2012 Nov;18(11):1630-8. doi: 10.1038/nm.2988. Epub 2012 Nov 7. PMID: 23135524; PMCID: PMC3616639.
[2] Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, McLaren S, Lin ML, McBride DJ, Varela I, Nik-Zainal S, Leroy C, Jia M, Menzies A, Butler AP, Teague JW, Quail MA, Burton J, Swerdlow H, Carter NP, Morsberger LA, Iacobuzio-Donahue C, Follows GA, Green AR, Flanagan AM, Stratton MR, Futreal PA, Campbell PJ. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011 Jan 7;144(1):27-40. doi: 10.1016/j.cell.2010.11.055.
[3] De Pagter M.S., van Roosmalen M.J., Baas A.F., Renkens I., Duran K.J., van Binsbergen E., Tavakoli-Yaraki M., Hochstenbach R., van der Veken L.T., Cuppen E., et al. Chromothripsis in Healthy Individuals Affects Multiple Protein-Coding Genes and Can Result in Severe Congenital Abnormalities in Offspring. Am. J. Hum. Genet. 2015;96:651–656. doi: 10.1016/j.ajhg.2015.02.005.
[4] Devereux TR, Risinger JI, Barrett JC. Mutations and altered expression of the human cancer genes: what they tell us about causes. IARC Sci Publ. 1999;(146):19-42.
[5] Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, McLaren S, Lin ML, McBride DJ, Varela I, Nik-Zainal S, Leroy C, Jia M, Menzies A, Butler AP, Teague JW, Quail MA, Burton J, Swerdlow H, Carter NP, Morsberger LA, Iacobuzio-Donahue C, Follows GA, Green AR, Flanagan AM, Stratton MR, Futreal PA, Campbell PJ. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011 Jan 7;144(1):27-40. doi: 10.1016/j.cell.2010.11.055.
[6] Pellestor, F. Chromoanagenesis: cataclysms behind complex chromosomal rearrangements. Mol Cytogenet 12, 6 (2019). https://doi.org/10.1186/s13039-019-0415-7
[7] Rasnic R, Linial M. Chromoanagenesis Landscape in 10,000 TCGA Patients. Cancers (Basel). 2021 Aug 20;13(16):4197. doi: 10.3390/cancers13164197.
[8] Suzuki E, Shima H, Toki M, Hanew K, Matsubara K, Kurahashi H, et al. Complex X-chromosomal rearrangements in two women with ovarian dysfunction: implications of chromothripsis/chromoanasynthesis-dependant and-independent origins of complex genomic alterations. Cytogenet Genom Res. 2016;150:86–92.
[9] Fukami M, Kurahashi H. Clinical consequences of chromothipsis and other catastrophic cellular events. Methods Mol Biol. 2018;1769:21–33.
[10] Wang K, Wang Y, Collins CC. Chromoplexy; a new paradigm in genome remodelling and evolution. Asian J Androl. 2013;15:711–2.
[11] Harper-Kirksey K. Erythroderma. Life-Threatening Rashes. 2018 Sep 12:265–77. doi: 10.1007/978-3-319-75623-3_19.
[12] Khaled A, Sellami A, Fazaa B, Kharfi M, Zeglaoui F, Kamoun MR. Acquired erythroderma in adults: a clinical and prognostic study. J Eur Acad Dermatol Venereol. 2010;24:781–8.
[13] Usatine RP, Smith MA, Chumley HS, Mayeaux EJ Jr. Erythroderma. In: Usatine RP, Smith MA, Chumley HS, Mayeaux Jr EJ, editors. The color atlas of family medicine. 2nd ed. New York: McGraw-Hill; 2013.
[14] Pelosof LC, Gerber DE. Paraneoplastic syndromes: an approach to diagnosis and treatment. Mayo Clin Proc. 2010 Sep;85(9):838-54. doi: 10.4065/mcp.2010.0099. Erratum in: Mayo Clin Proc. 2011 Apr;86(4):364.
[15] Kaaks R, Sookthai D, Łuczyńska A, Oakes CC, Becker S, Johnson T, Johansson A, Melin B, Sjöberg K, Trichopoulos D, Trichopoulou A, Lagiou P, Mattiello A, Tumino R, Masala G, Agnoli C, Boeing H, Aleksandrova K, Brennan P, Franceschi S, Roulland S, Casabonne D, de Sanjose S, Sánchez MJ, Huerta JM, Ardanaz E, Sala N, Overvad K, Tjønneland A, Halkjær J, Weiderpass E, Bueno-de-Mesquita HB, Vermeulen R, Peeters PH, Vineis P, Kelly RS, Khaw KT, Travis RC, Key TJ, Riboli E, Nieters A. Lag times between lymphoproliferative disorder and clinical diagnosis of chronic lymphocytic leukemia: a prospective analysis using plasma soluble CD23. Cancer Epidemiol Biomarkers Prev. 2015 Mar;24(3):538-45. doi: 10.1158/1055-9965.EPI-14-1107. Epub 2014 Dec 26.
[16] Nilius-Eliliwi V, Gerding WM, Schroers R, Nguyen HP, Vangala DB. Optical Genome Mapping for Cytogenetic Diagnostics in AML. Cancers (Basel). 2023 Mar 9;15(6):1684. doi: 10.3390/cancers15061684.
[17] Hernández Guillamet G, Morancho Pallaruelo AN, Miró Mezquita L, Miralles R, Mas MÀ, Ulldemolins Papaseit MJ, Estrada Cuxart O, López Seguí F. Machine Learning Model for Predicting Mortality Risk in Patients With Complex Chronic Conditions: Retrospective Analysis. Online J Public Health Inform. 2023 Dec 28;15:e52782. doi: 10.2196/52782.
